



2. 中国科学院微生物研究所,北京 100101
2. Institute of Microbiology,Chinese Academy of Sciences,Beijing 100101
葡萄糖氧化酶(Glucose oxidase,EC 1.1.3.4,简称GOD)是氧化-还原酶类的典型代表,能够在氧气存在时专一催化β-D-葡萄糖生成葡萄糖酸和过氧化氢。因其氧化反应生成过氧化氢,在实际应用中葡萄糖氧化酶被用作抗菌剂。另外,葡萄糖氧化酶作为生物催化剂常用于临床诊断、食品和饮料保鲜[1];葡萄糖氧化酶作为酶电极还可用于生物传感器领域[2]。
葡萄糖氧化酶广泛分布于动植物及微生物体内。研究及生产葡萄糖氧化酶的主要菌株为黑曲霉(Abperrillus niger)和点青霉(Penicillium notatum),这是因为霉菌产酶能力强,易于规模化生成;但黑曲霉和青霉菌发酵生产GOD过程中,过氧化氢酶、纤维素酶及淀粉酶等大量杂酶的存在给纯化带来相当大的困难[3]。
重组葡萄糖氧化酶基因进行异源表达可有效地解决这些问题[4, 5]。尤其是毕赤酵母表达外源蛋白具有表达量高、稳定性好、培养成本低和产物易分离纯化等优点,适于大体积高密度连续发酵,具有强且易控的醇氧化酶(Alcohol oxidase,AOX)启动子等优点[6],可严格控制外源基因的表达[7]。
近年来,河北省微生物研究所对保存的青霉属的点青霉No.8312菌株,采用紫外线诱变方法处理,筛选到了产酶较高菌株,本研究从点青霉菌中克隆得到葡萄糖氧化酶基因,并将其构建在 pMD-AOX载体上,转化毕赤酵母X33,进行点青霉菌的GOD基因实现异源高效表达。同时,对重组表达的葡萄糖氧化酶进行纯化,并对其性质进行初步研究。
1 材料与方法 1.1 材料 1.1.1 菌株和质粒点青霉Penicillium notatum菌株和Escherichia coli DH5α菌株由河北省微生物研究所保存。毕赤酵母Pichia pastoris X33由中科院微生物所馈赠。毕赤酵母表达载体 pMD-AOX(含AOX强启动子和终止子,G418抗性基因)为中科院微生物所构建。
1.1.2 主要试剂限制内切酶Not Ⅰ、Pme Ⅰ和EcoR Ⅰ以及Q5 DNA聚合酶为NEB公司产品;邻联茴香胺、辣根过氧化物酶、牛血清白蛋白等购自Sigma公司;蛋白质浓度测定试剂盒为上海生工公司产品;DNA Marker和蛋白Marker 购于Fermentas 公司;其他均为国产分析纯试剂。真菌基因组提取试剂盒购于北京索莱宝科技有限公司;质粒提取试剂盒、普通DNA 回收试剂盒和琼脂糖凝胶回收试剂盒购于天根生化科技(北京)有限公司。PCR 扩增引物由上海生工合成。
1.1.3 培养基察氏培养基、含有Amp抗性的 LB培养基、含有G418 的YPD培养基分别用以培养点青霉、大肠杆菌和酵母菌,BMGY培养基(酵母提取物1%、蛋白胨2%、pH6.0磷酸钾缓冲液100 mmol/L、YNB 1.34%、生物素4×10-5%、甘油1%)为毕赤酵母甲醇诱导表达前培养基。YPCS发酵培养基(g/L)(酵母提取物10,蛋白胨20,酪蛋白水解物5,山梨醇5)为毕赤酵母诱导表达培养基。诱导培养时,每24 h加入1%的甲醇。
1.2 方法 1.2.1 点青霉基因组的提取按照真菌基因组提取试剂盒方法提取P. notatum的基因组DNA。
1.2.2 葡萄糖氧化酶基因的扩增扩增来自于P. notatum的葡萄糖氧化酶引物的设计参照NCBI上报导的葡萄糖氧化酶序列(GenBank 登录号:XM_002563405.1和JN809249.1),其5’端引物序列P1为:5’-ATGAAGTCCACTATTATCACCTCCA-3’,其3’端引物序列P2为:5’-CTAGGCACTTTTGGCA-TAGTCTTCA-3’,用这对引物以P. notatum基因组为模板进行PCR扩增。反应体系为50 μL:基因组DNA 100 ng,引物各0.2 μmol/L,dNTPs 250 μmol/L,5×Q5 High-Fidelity DNA Polymerase Buffer 10 μL,Q5 High-Fidelity DNA Polymerase 0.5 μL。PCR反应条件:98℃ 5 min;98℃ 30 s,65℃ 30 s,72℃ 60 s,共30个循环;72℃ 10 min。PCR产物经DNA回收试剂盒回收。将经鉴定无误的连有点青霉葡萄糖氧化酶基因的重组质粒进行测序。
1.2.3 葡萄糖氧化酶基因重组表达载体的构建为了将葡萄糖氧化酶基因在毕赤酵母中表达,首先去掉其自身的信号肽编码序列,通过PCR方法设计引物将点青霉的葡萄糖氧化酶基因序列中的信号肽编码序列去除。引物序列如下:GodF:5’-GGCTGAAGCTTACGTAGAATTCTATAGCCCGGCCGAGCAGATCGAC-3’;GodR:5’-GAGATGAGTTTTTGTTCTAGAGCGGCCGCCTAAGCTTTCTTGGCATAGTCTTC-3’(下划线分别为上下游引物中限制性内切酶EcoR Ⅰ和Not Ⅰ识别位点)。PCR产物经纯化回收采用GIBSON连接到表达载体 pMD-AOX的EcoR Ⅰ和Not Ⅰ位点上,转化到 E. coli DH5α。酶切验证筛选阳性克隆,并送测序。
1.2.4 毕赤酵母的转化及诱导培养采用电转化法[8],重组质粒 pMD-AOX-GOD经Pme Ⅰ酶切后线性化。用 2 μg线性化 pMD-AOX-GOD质粒电击转化80 μL毕赤酵母X33感受态细胞,然后将转化细胞涂于含 100 μg/mL G418的YPD平板上,30℃培养96 h。随机挑选阳性菌落转接液体YPCS(含100 μg/mL G418,下同),置于 30℃、200 r/min条件下振荡培养3 d,期间每隔24 h加1%的甲醇。
1.2.5 10 L发酵罐发酵选取摇床水平上葡萄糖氧化酶活性最高的转化子研究其发酵罐条件下葡萄糖氧化酶的表达。挑单克隆接种于YPD培养基培养10 h,按3%接种量转接于100 mL BMGY 培养基至OD600≈6接种于发酵罐,装液量为2 L BSM培养基,灭菌后,以10%发酵体积接种。参照酵母表达手册,以浓氨水控制pH5,培养温度控制在30℃。培养阶段甘油耗尽时(溶氧值DO开始明显上升),开始流加50%甘油,至OD600≈180,停止流加甘油,4 h后往培养基中甘油耗尽,开始流加甲醇诱导,期间每隔24 h加维生素C水溶液(终浓度80 μg/L),12 h测定发酵液酶活。发酵结束以6 000×g、4℃离心20 min收集发酵液上清,将所有收集到的上清液用超滤浓缩纯化,提高分泌蛋白的浓度和纯度,用BCA 法测蛋白浓度。用上清液进行10%聚丙烯酰胺凝胶电泳,考马斯亮蓝R-250染色,脱色液(甲醇250 mL∶冰醋酸50 mL∶水200 mL)脱色,观察GOD蛋白的表达。
1.2.6 葡萄糖氧化酶的酶活和比活测定 1.2.6.1 GOD活性测定一般采用邻联茴香胺分光光度法。在3 mL的反应体系中,包含0.5 mol/L醋酸缓冲液(pH5.1),0.17 mmol/L邻联茴香胺溶液和1.72%葡萄糖溶液及0.1 mg/mL辣根过氧化物酶,35℃震荡混匀,加入0.1 mL葡萄糖氧化酶溶液,用分光光度计,于A=500 nm自动记录随时间变化的吸光度值,根据公式,计算葡萄糖氧化酶活力单位。
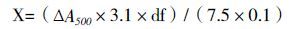
式中,X:样品酶活(U/ mL);ΔA500:活度值;3.1:反应体积;df为稀释倍数;7.5:葡萄糖内酯A500消光系数;0.1:加入酶液体积。
1.2.6.2 葡萄糖氧化酶比活性的测定蛋白浓度测定采用上海生工Modified Bradford Protein Assay Kit 测定。得到蛋白质含量以后,同时测得其葡萄糖氧化酶的活性,由此得到葡萄糖氧化酶的比活性。
1.2.7 酶的分离纯化取发酵液在4℃、6 000 r/min离心20 min,去除发酵液中的菌体;将离心得到的上清液加入无水乙醇(终浓度20%),4℃静置过夜,6 000 r/min离心20 min;取上清液,再加入无水乙醇(终浓度60%),4℃静置过夜,6 000 r/min离心20 min,将得到的沉淀用0.1 mol/L pH7.4的磷酸盐缓冲溶液稀释到合适的酶活即得到初步纯化的GOD酶液。将初步纯化的GOD酶液采用DEAE-Sephadex A50进一步纯化后经超滤浓缩后即可得精纯度的葡萄糖氧化酶酶液。
1.2.8 葡萄糖氧化酶的酶学性质研究 1.2.8.1 温度对酶活力的影响(1)最适温度:取10 μL一定浓度的纯化后酶液,分别于35℃、40℃、45℃、50℃、55℃、60℃、65℃和70℃下测定其酶活力。(2)温度耐受:取一定量的纯化后的酶液,分别于35℃、40℃、45℃、50℃、55℃、60℃和65℃孵育30、60、90和120 min测定酶活力。
1.2.8.2 pH对酶活力的影响(1)最适pH:取10 μL一定浓度的纯化后酶液,分别于不同pH的缓冲液(终浓度均为 40 mmol/L)中,40℃测定其酶活力,并计算其比活。(2)pH耐受:取一定量的纯化后酶液,分别于不同pH的缓冲液中孵育5 h;然后,于 40℃下,取一定浓度的 10 μL 酶液测定酶活力。
1.2.8.3 金属离子对酶活力的影响将10 μL一定浓度的纯化后酶液,在 pH6.0缓冲液混合后,分别加入终浓度为 0.1、1.0和10.0 mmol/L的金属离子,补水到125 μL,40℃下测定其酶活力。
2 结果 2.1 点青霉GOD基因的克隆分析以点青霉基因组DNA为模板,引物P1和P2进行PCR扩增(图 1)。序列分析结果表明,其基因组DNA长1 815 bp,GC含量为55.4%,无内含子,编码605个氨基酸(图 2)。用BLAST 进行同源分析,结果显示与产黄青霉(Penicillium chrysogenum)的GOD基因(GenBank登录号:XM_002563405.1)有99% 的核酸序列相似性。推导的GOD氨基酸序列与已知的产黄青霉氨基酸序列相似性达99.8%,仅第15位氨基酸发生了Thr-Ala的转变。

|
| 图 1 点青霉基因组PCR扩增产物琼脂糖凝胶电泳 M:DNA Marker;1:PCR 扩增产物 |

|
| 图 2 GOD氨基酸同源性比较 |
将经引物GodF和GodR进行PCR扩增并纯化回收的点青霉GOD基因和经过EcoR Ⅰ和Not Ⅰ双酶切的载体pMD-AOX进行Gibson连接以构建重组质粒。用重组质粒转化感受态大肠杆菌DH5α,并在含Amp的LB平板上筛选阳性菌落。阳性菌落质粒经EcoR Ⅰ和Not Ⅰ双酶切后,得到约6.5 kb和1.8 kb的两个DNA片段,亦与预期大小相符(图 3)。重组质粒测序结果显示目的基因的完整阅读框序列已正确插入到pMD-AOX载体α-因子信号肽DNA序列的下游,表明含点青霉GOD基因的毕赤酵母表达载体 pMD-AOX-GOD已构建成功。

|
| 图 3 重组质粒pMD-AOX-GOD琼脂糖凝胶电泳鉴定 M:DNA Marker;1-5:重组质粒pMD-AOX-GOD 经EcoR Ⅰ和Not Ⅰ双酶切的产物 |
将重组的表达质粒 pMD-AOX-GOD经Pme Ⅰ酶切后线性化后,电转化毕赤酵母X33感受态,在YPD平板生长3 d后,随机挑选阳性菌落转接液体YPCS,经甲醇诱导72 h后,测定发酵液上清中的葡萄糖氧化酶活性,从32个转化子中挑选出6株有葡萄糖氧化酶活性的转化子,表达率为16.7%。说明表达载体pMD-AOX-GOD 于毕赤酵母X33中得到有功能活性表达。
2.4 10 L发酵罐发酵结果选取X33-GOD中酶活最高株为实验株在10 L发酵罐中发酵,在甲醇未诱导之前,处于菌株培养和碳源饲喂阶段,在这两个阶段内,菌体大量增长,此时无葡萄糖氧化酶蛋白的表达。随着甲醇的诱导,发酵液中的葡萄糖氧化酶活性逐渐增加(图 4),诱导144 h后发酵液中GOD活性达到最高496 U/mL,蛋白质表达量达到9.4 g/L,SDS-PAGE(图 5)结果表明,发酵液中葡萄糖氧化酶蛋白表达量也在不断积累,发酵液上清中GOD含量非常高,可以达到电泳纯,可大大降低后期GOD纯化的成本。

|
| 图 4 葡萄糖氧化酶发酵液酶活增长曲线 |

|
| 图 5 10 L发酵罐中葡萄糖氧化酶经甲醇诱导不同时间内的表达量 M:蛋白Marker;1-12:经甲醇诱导0、24、36、48、60、72、84、96、108、120、132和144 h葡萄糖氧化酶表达量 |
取 300 mL发酵液经高速冷冻离心,酒精沉淀、溶解后,上DEAE-Sephadex A50层析柱,超滤浓缩,收率可达94%。纯化前后电泳图谱(图 6)显示,在甲醇的诱导下,GOD在X33中的表达量明显高于其他杂蛋白。发酵液上清经分离纯化后,可见一条浓染的约90 kD的蛋白质条带,点青霉GOD理论分子量为60.2 kD,这与图 4中点青霉GOD电泳结果(65 kD)相似,但是重组酵母 GOD蛋白条带明显比点青霉GOD分子量大。根据该基因中有潜在的糖基化位点,毕赤酵母表达系统又有转录翻译后的加工功能,推测毕赤酵母表达的GOD蛋白质为糖蛋白。
通过蛋白含量标准曲线测定出酶液中的蛋白含量,再通过常规的葡萄糖氧化酶酶活性测定方法,最后得到了该酶的比活。经过测定,毕赤酵母X33-GOD比活为123.0 U/mg。同时,测定的青霉GOD比活仅为101.3 U/mg。

|
| 图 6 重组表达GOD蛋白SDS-PAGE M:蛋白Marker;1:毕赤酵母重组表达蛋白质上清液;2:浓缩纯化后的毕赤酵母重组表达蛋白;3:点青霉表达GOD |
在pH6.0的测活条件下,改变测活条件的反应温度,研究温度对酶活力的影响,最适温度(图 7-A)显示,该酶催化反应的最适温度为40℃-45℃,70℃下酶活力基本接近0。重组 GOD对温度的稳定性(图 7-B)显示,在35℃下孵育90 min酶活力基本保持不变,孵育到120 min时降低了12个酶活单位;在40℃下孵育90 min降低了15个酶活单位,孵育到120 min时降低了20个酶活单位;在45℃和50下℃孵育30 min降低了15个酶活单位,孵育到120 min时降低了40个酶活单位;在55℃下孵育30 min降低了20个酶活单位,孵育到120 min时降低了50个酶活单位;60℃孵育30 min,酶活力大约降低了30个酶活单位,孵育90 min后酶活力几乎检测不到。

|
| 图 7 温度对GOD酶活力(A)及热稳定性(B)的影响 |
在40℃的测活体系中,改变缓冲液的pH值,分析重组表达的葡萄糖氧化酶的酶活力发现,酶活力对反应的pH作图,得到一个相对平缓的曲线,最适 pH为6.0(图 8-A)。重组葡萄糖氧化酶对pH的稳定性(图 8-B)表明,在不同pH的缓冲液中孵育5 h,重组表达的葡萄糖氧化酶在pH3.5-7.0的缓冲液中的酶活力能维持在90%以上,在pH3.0和pH8.0缓冲液中,酶的活性降低为原来的60%左右。

|
| 图 8 pH对GOD酶活力(A)及pH稳定性(B)的影响 |
不同浓度的金属离子对葡萄糖氧化酶的酶活力影响(图 9)显示,在0.1 mmol/L的金属离子浓度条件下,葡萄糖氧化酶的酶活力大多能维持在80%以上(除Ag+),Ag+对其有抑制作用;在金属离子浓度为1.0 mmol/L时,葡萄糖氧化酶的酶活力大多能维持在80%以上,其中Zn2+对其有激活作用,Ag+对其有抑制作用;当金属离子浓度为10 mmol/L时,Ag+、Cu2+和Fe3+对GOD活力的抑制作用非常明显,Ag+尤为显著,GOD的剩余酶活力仅为1%。

|
| 图 9 金属离子对重组GOD的影响 |
GOD作为安全的生物抗氧化剂,在科研医药和食品工业生产上被广泛应用。目前,在产生菌的诱变育种筛选及产酶条件的优化上做了大量工作[9-12],也有一些学者进行了黑曲霉的葡萄糖氧化酶基因在毕赤酵母中的克隆和表达[3, 13, 14]。毕赤酵母表达系统优点是蛋白表达量高,可将外源蛋白分泌至胞外,且被表达的蛋白中杂蛋白少;毕赤酵母还具有对表达产物进行加工、折叠和翻译后修饰[15]。本研究依据已知产黄青霉的GOD序列从点青霉中成功克隆了GOD基因,通过BLAST,相似性达99%,说明在青霉属中GOD相对保守;点青霉GOD发生了氨基酸置换(Thr15-Ala),由于点突变能改变酶分子的催化反应动力学参数及催化反应活化能的现象,从而影响蛋白质热稳定性[16]。
本研究构建的毕赤酵母菌工程菌株X33-GOD在10 L发酵罐中30℃、pH6.5条件下发酵培养时,培养液GOD 酶活可达496 U/mL。X33-GOD比原始出发菌株点青霉的酶活(30 U/mL)提高近16倍。郭元芳等[13]报道的毕赤酵母SMD1168 表达黑曲霉accc30161GOD,其培养液上清GOD酶活力为107.18 U/mL;周亚凤等[3]构建出GOD的高产酵母工程菌株,经甲醇诱导3-4 d,发酵液中的GOD 活力为30-40 U/mL;李卓夫[17]将青霉GOD在毕赤酵母GS115中异源表达,在3 L发酵罐水平中,甲醇诱导132 h,发酵液中的最高酶活为111.3 U/mL。因此,本研究构建的毕赤酵母X33-GOD工程菌株具有很高的推广应用价值。
本研究所分泌表达的GOD的单亚基分子量为90.0 kD,比原始菌株点青霉所产GOD单亚基分子量高约25 kD,分子量差异主要是由于糖基化造成的。糖基化对毕赤酵母分泌表达的GOD在蛋白折叠中起重要作用,能使酶蛋白结构具有更好的稳定性,这种过糖基化现象在黑曲霉GOD在毕赤酵母的表达中也存在[3]。
在青霉和黑曲霉中提取GOD需要经过DEAE离子交换层析和凝胶过滤层析两步才能纯化,花费时间长并且费用也相对较高[18, 19]。毕赤酵母发酵表达GOD,不需破碎菌体,发酵的上清液经离心就能得到,且毕赤酵母表达系统极少分泌自身蛋白,杂蛋白含量低,只需经过酒精沉淀和一步离子交换层析就可以达到电泳纯,纯化收率高,费用低廉,该方法适合工业化生产,有利于大规模纯化。
酶学性质研究发现,相比以往报道的重组黑曲霉GOD和产黄青霉GOD的最适温度仅有40℃[19, 20],本研究重组表达的来源于点青霉的GOD的最适温度是40-45℃,范围较宽;并且该酶热稳定性良好,在35-55℃之间都能保持良好的酶学活性,酶的热稳定性提高很可能也是由于过糖基化所造成的,毕赤酵母分泌表达的黑曲霉GOD也表现出了热稳定性的提高[21];其次该酶在pH3.5-7.0之间能保持90%以上酶相对活力,有较高的应用价值。
本研究中Zn2+对酶有一定的激活作用,而Ag+、Cu2+和Fe3+ 对酶的活性有抑制作用,这与张茜[19]报道的尼崎青霉菌GOD的性质一样。而Kelley等[22]报道当Cu2+ 达到10 mmol/L时,GOD活性仍没有被抑制;苏茉等[18]研究认为Zn2+对黑曲霉H1-9b所产GOD的活性有抑制作用。
4 结论将点青霉菌的葡萄糖氧化酶基因(GOD)克隆到载体 pMD-AOX质粒,线性化质粒pMD-AOX-GOD,转化到毕赤酵母Pichia pastoris X33中异源表达,获得了GOD的高产毕赤酵母工程菌株。结果显示,与点青霉GOD 相比,毕赤酵母Pichia pastoris X33GOD具有更高的发酵酶活和比活。
| [1] | Wong CM, Wong KH, Chen XD. Glucose oxidase:natural occurrence,function,properties and industrial applications[J]. Appl Microbiol Biotechnol, 2008, 78(6) : 927–938. |
| [2] | Yin B, Yuan R, Chai Y, et al. Amperometric glucose biosensors based on layer-by-layer assembly of chitosan and glucose oxidase on the Prussian blue-modified gold electrode[J]. Biotechnol Lett, 2008, 30(2) : 317–322. |
| [3] | 周亚凤, 张先恩, 刘虹, 等. 黑曲霉葡萄糖氧化酶基因的克隆及其在酵母中的高效表达[J]. 生物工程学报, 2001, 17(4) : 400–405. |
| [4] | Damasceno L, Huang CJ, Batt C. Protein secretion in Pichia pastoris and advances in protein production[J]. Appl Microbiol Biotechnol, 2012, 93(1) : 31–39. |
| [5] | Kombrink A. Heterologous production of fungal effectors in Pichia pastoris[M]. Plant Fungal Pathogens: Humana Press, 2012 : 209-217. |
| [6] | Krainer F, Dietzsch C, Hajek T, et al. Recombinant protein expression in Pichia pastoris strains with an engineered methanol utilization pathway[J]. Microb Cell Fact, 2012, 11(1) : 22–35. |
| [7] | 闵兆升, 郭会明, 颜旭, 等. 巴斯德毕赤酵母(P. pastoris)高密度发酵研究进展[J]. 生物技术通报, 2014(3) : 42–49. |
| [8] | Gietz RD, Woods RA. Genetic transformation of yeast[J]. Biotechniques, 2001, 30(4) : 816–820. |
| [9] | 朱运平, 褚文丹, 李秀婷, 等. 产胞外葡萄糖氧化酶菌株的分离鉴定及其诱变育种研究[J]. 中国食品学报, 2014, 14(4) : 95–103. |
| [10] | 唐菁, 赵芯, 苏茉, 等. 黑曲霉H1-9b发酵产葡萄糖氧化酶的工艺优化[J]. 食品科学, 2013, 34(9) : 220–223. |
| [11] | 胡常英, 刘丽娜, 王云鹏, 等. 点青霉高产葡萄糖氧化酶菌株的诱变选育[J]. 河北省科学院学报, 2006, 23(3) : 11–15. |
| [12] | 梁静娟, 李筱瑜, 官威, 等. 产葡萄糖氧化酶黑曲霉的诱变选育及葡萄糖酸钙发酵条件的研究[J]. 食品工业科技, 2010, 31(12) : 218–220. |
| [13] | 郭元芳, 孙高英, 郝建荣, 等. 黑曲霉葡萄糖氧化酶在毕赤酵母SMD1168中的表达[J]. 生物技术, 2013, 23(4) : 25–29. |
| [14] | Gu L, Zhang J, Liu B, et al. High-level extracellular production of glucose oxidase by recombinant Pichia pastoris using a combined strategy[J]. Appl Biochem Biotech, 2015, 175(3) : 1429–1447. |
| [15] | 陆永超, 蒋琳. 毕赤酵母高效表达策略概述[J]. 微生物学免疫学进展, 2013, 41(1) : 70–76. |
| [16] | 袁有忠, 赵思汇, 周宗祥, 等. 一个耐热碱性磷酸酯酶突变型的研究[J]. 遗传学报, 2000, 27(4) : 361–368. |
| [17] | 李卓夫. 葡萄糖氧化酶基因的克隆及高效表达[D]. 长春:长春理工大学,2008. |
| [18] | 苏茉, 高亚朋, 梁建荣, 等. 黑曲霉H1-9b葡萄糖氧化酶的分离纯化及部分性质研究[J]. 食品科学, 2011, 32(3) : 181–185. |
| [19] | 张茜, 傅婉辉, 康劲翮, 等. 青霉葡萄糖氧化酶的分离纯化及性质研究[J]. 厦门大学学报:自然科学版, 2009, 48(1) : 99–102. |
| [20] | 郝杰清, 王帅坤, 师慧, 等. 重组毕赤酵母葡萄糖氧化酶的纯化和性质[J]. 食品科学, 2013, 34(9) : 159–163. |
| [21] | 黄亮. 黑曲霉A9葡萄糖氧化酶基因克隆及其在毕赤酵母中的表达[D]. 保定:河北农业大学,2007. |
| [22] | Kelley RL, Reddy CA. Purification and characterization of glucose oxidase from ligninolytic cultures of Phanerochaete chrysosporium[J]. J Bacteriol, 1986, 166(1) : 269–274. |