




2.石河子大学动物科技学院,石河子 832003
2. College of Animal Science and Technology,Shihezi University,Shihezi 832003
基因突变已成为当今生命科学研究的热点之一,其检测方法也随之迅速发展,变性高效液相色谱分析,单链构象异构多态分析技术,毛细管电泳,直接测序,变性梯度凝胶电泳与化学或酶促裂解等突变检测方法中,酶错配切割( Enzyme mismatch cleavage,EMC)方法临床应用简单,具有一定的优越性。酶错配切割的原理为利用错配分辨酶识别错配,插入和缺失切割错配的DNA,异源双链DNA被识别错配的核酸酶识别并切割成DNA片段[1],正常DNA和突变DNA在一起通过聚合酶链反应(PCR)变形后再缓慢复性,同时形成同源双链(Homoduplex,HO)和异源双链(Heteroduplex,HE)。EMC中Surveyor 核酸酶是CELI家族成员,对DNA错配部位有高度特异性,识别异源双链DNA中的错配碱基的位置并在错配扭曲链两端的3'处切断,能准确切割异源双链DNA错配位点[2];T7 Endonuclease I(T7EI)作用于双链 DNA,沿 5'→3'方向催化去除 5'单核苷酸,它既能从 5'末端起始消化,也能从双链 DNA 的切刻或缺口处起始消化异源双链DNA序列[3]。高分辨率熔解曲线分析技术(High resolution melting,HRM)是近年来兴起的一种检测基因突变、进行基因分型和SNP检测的新工具,可以迅速检测出核酸片段中单碱基的突变[4]。HRM技术自2002年应用于基因分型、突变扫描、序列匹配检测以来,受到了高度重视并快速发展。HRM 技术因其速度快,操作简便,高通量,灵敏性特异性高,对样品无污染等优点而被迅速应用在生命科学、医学、农学及畜牧业等领域的研究工作中[5, 6]。
三种检测方法都以变性退火突变和野生型DNA序列,形成扭曲的双螺旋DNA(Distorted duplex DNA)为基础。Surveyor nuclease和T7EI利用核酸内切酶特异性切割扭曲的双螺旋DNA,经电泳显示的DNA条带图谱,确定是否产生突变。并根据酶切电泳获得DNA条带的光密度值,计算确定突变和野生型DNA序列的比率。HRM法是利用扭曲的双螺旋DNA影响饱和染料结合量,荧光信号经高灵敏度信号仪收集后,通过检测荧光信号变化,绘制溶解曲线,通过熔解曲线变化,确定是否存在突变型基因。实验采用3种人工核酸酶生物学活性检测方法确定目标位点是否发生突变(图 1),通过比较分析得出3种检测方法的优缺点,旨在为实验室分析确定细胞利用非同源末端连接修复DNA双链断裂结果提供参考。

|
| 图 1 三种检测内源性基因修饰方法的示意图 |
Surveyor nuclease试剂盒(SURVEYOR Mutation Detection For Standard GelElectrophroesis),TRANSG-ENOMI公司;T7EI试剂盒,New England Biolabs公司;LightCycler®480High Resolution Melting Master,Ro-che公司;QuickExtract DNA Extraction solutio1.0(基因组 DNA 快速抽提液),Epicentre公司;Nanophot-ometer微量分光光度计,德国Implen公司;100 bp DNA Marker,北京全式金生物技术有限公司。
1.2 方法 1.2.1 Cas9和TALEN作用靶点确定实验设计Cas9作用于MSTN基因第一外显子,TALEN作用于MSTN基因第三外显子。Cas9和TALENs质粒由广州复能基因有限公司合成(图 2)。

|
| 图 2 Cas9(A)和TALEN(B)作用位点及Surveyor、T7E1酶切、HRM引物序列 |
将萨福克绵羊成纤维细胞电转染,采用自己配制的电转液100 µL,CZ-167电转程序,质粒总浓度4 µg,2×106个细胞,电击后37℃静置10 min,加到6孔板内培养,电转3组细胞:Cas9质粒作用于MSTN基因第一外显子;TALEN质粒作用于MSTN基因第三外显子;Pmax绿色荧光蛋白表达质粒作用对照组细胞。培养电转染细胞72 h,收集实验组及对照组细胞。QuickExtract DNA提取液裂解细胞提取细胞基因组,68℃ 15 min;95℃ 8 min处理细胞裂解液,-20℃保存备用。
1.2.3 引物设计通过NCBI查找绵羊MSTN基因全序列(Sequence ID: gb|DQ530260.1|),设计MSTN第一和第三外显子Surveyor 核酸酶、T7E1和HRM检测基因突变的PCR引物(Surveyor nuclease和T7E1引物为SVF/R,HRM引物为HRMF/R)(表 1)。

Surveyor核酸酶和T7EI酶切分析的PCR产物需要高保真热启动酶扩增,保证扩增片段的特异性。TALEN和Cas9靶点周边序列PCR扩增反应体系(表 2)。PCR反应程序:95℃ 3 min;95℃ 20 s,60℃ 30 s,72℃ 30 s,共35个循环;72℃ 5 min;4℃保存。琼脂糖凝胶纯化PCR扩增产物,使用Nanopho-tometer微量分光光度计测胶回收产物浓度。
核酸酶酶切PCR纯化产物 由于非同源末端连接(NHEJ)修复机制,经TALEN,Cas9修饰的基因组DNA在TALEN,Cas9靶位点附近的几个碱基序列发生突变。对于Surveyor和T7EI错配检测,首先对PCR扩增产物再一次变性和退火以形成 DNA 异源双链。由于样品中同时存在TALEN,Cas9修饰以及未修饰的DNA,异源双链中可能包括TALEN,Cas9修饰过的DNA的一条链和未修饰的DNA的一条链,也有可能由两个不同的经TALEN,CAS9修饰过的 DNA再退火形成。变性DNA双链经缓慢退火复性形成异源双链。使用400 ng纯化的PCR产物退火处理。PCR产物退火体系20 µL:Herculase II reaction buffer,5×4 µL;PCR纯化产物×(400 ng);ddH2O补加到20 µL。样品混匀、离心后,进行退火处理。使用以下程序(表 3)进行退火反应。
为了确定CRISPR/Cas9和TALEN的切割效率,用 Surveyor 核酸酶S处理交叉杂交形成的异源双链和同源双链。Surveyor核酸酶S消化体系20 μL:MgCl2 solution,0.15 mol/L 2 µL;Surveyor nuclease S 1 µL;Surveyor enhancer S 1 µL;退火产物16 µL。将PCR管放在42℃水浴锅恒温孵育1 h,然后加入2 µL Stop solution终止消化,储存于-20℃冰箱备用。
1.2.6 T7EI处理PCR胶回收产物400 ng纯化的PCR产物退火处理,样品添加同1.2.5。T7EI核酸酶消化退火产物样品混匀、离心后,进行退火处理同表 4。T7EI核酸酶处理PCR纯化产物20 μL:T7 Endonuclease 0.5 µL;NEBuffer 2 10×0.2 µL;ddH2O 1.3 µL;退火产物18 µL。样品混匀离心后,将PCR管放在37℃水浴锅恒温孵育1 h,-20℃冰箱备用。
8%的PAGE检测Surveyor核酸酶S和T7EI消化退火产物Cas9和TALEN切割效果。Surveyor核酸酶S消化产物加6×Loading buffer 4 µL上样即可,T7EI核酸酶消化产物需要添加EDTA,使其终浓度为45 mmol/L,也可以通过添加6.94 µL的混合物:0.5 mol/L的EDTA与6×xylene cyanol loading dye比例2.45∶4.49[7]。酶切结果使用quantity one定量软件分析。
1.2.8 HRM检测根据DNA序列的长度,GC含量以及碱基互补性差异,应用高分辨率的熔解曲线对样品进行分析,其分辨精度可以达到对单个碱基差异的区分[8]。HRM利用特定的染料可以插入DNA双链中的特性,从而对样品进行检测。基于这种检测原理,HRM检测不受突变碱基位点和种类的局限,既可以对未知突变进行筛查、扫描,又可以对已知突变进行分析,亦可用于短片段重复序列的分析,所需要的只是在常规PCR基础上增加一个饱和染料,常规PCR后不需电泳等后处理。
按照LightCycler®480High Resolution Melting Mas-ter说明书要求每个样品3份,每份20 µL体系(表 4)混匀后加到96孔板内,LightCycler®480 II型仪器设定相应PCR反应程序,结果使用LightCycler®480基因分型软件1.5版本分析。高分辨率的熔解分析方法分析方便,因为不需要处理步骤和分离步骤[9]。
1.3 非同源末端连接百分比计算公式ƒcut=(Cleavage Band1+Cleavage Band2)/(Uncl-eaved Band+Cleavage Band1+Cleavage Band2)
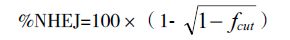
Cas9和TALEN分别作用于MSTN基因的第一外显子和第三外显子,在作用位点处设计Surveyor,T7EI核酸酶检测以及HRM检测的引物(图 3)。

|
| 图 3 PCR扩增Cas9(A)、TALEN(B)作用于MSTN基因示意图 |
电转染9 h细胞换液,72 h荧光显微镜下观察电转染的效率(图 4)。

|
| 图 4 Pmax绿色荧光蛋白表达质粒电转染结果图 |
PAGE胶检测Surveyor和T7EI酶切效果,并使用quantity one定量软件分析,定量结果使用非同源末端连接百分比计算公式计算NHEJ百分比(图 5)。

|
| 图 5 CRISPR/Cas 9和TALEN的活性酶切鉴定图 |
第一外显子Surveyor和T7EI 检测引物扩增长度为575 bp,Cas9的作用靶点在243-262 bp处,Surveyor和T7EI 在作用靶点发生酶切作用,切开的条带大小为243 bp和313 bp左右,但是在MSTN第一外显子的161 bp处产生一个突变位点,实验将未电转染的萨福克绵羊成纤维细胞和电转染CRISPR/Cas9的细胞提取基因组,分别使用MSTNE1-F/R引物扩增,将其胶回收产物测序,结果均为在161 bp处存在SNP位点(图 6)。Surveyor核酸酶检测到这一突变位点并将其切开,两个突变位点可产生条带大小243 bp和313 bp,161 bp和414 bp左右,但是414 bp这个中间产物中还存在Cas9的突变位点,会产生100 bp和314 bp左右的条带,酶切产生6条带,分别为100 bp,161 bp,243 bp,313 bp,314 bp和414 bp,313 bp和314 bp左右的条带在酶切图上只产生了一条带,所以酶切图上只产生了5条带切开的条带;T7EI只切开了Cas9的酶切位点,产生了两条切开的条带。MSTN第三外显子Surveyor和T7EI 检测引物扩增长度为532 bp,TALEN的作用靶点在239-255 bp处,Surveyor和T7EI在作用靶点发生酶切作用,切开的条带大小为277 bp和293 bp左右。

|
| 图 6 MSTN第一外显子基因突变位点测序结果图 |
样品基因型的分析结果见图 7。

|
| 图 7 HRM检测Cas9和TALEN基因分型图 |
在基因组编辑技术的推动下,建立动物和细胞模型的速度也越来越快。在这种情况下,基因分型过程成为了新的瓶颈[10, 11]。目前,检测基因靶位点突变的技术主要有Surveyor核酸酶、T7E1分析和高分辨率熔解分析(HRMA)3种,这3种检测方法简单、方便、分析更快,为最常用的检测突变方法。
Surveyor 和T7E1 能有效地切开不匹配的位点,切割的片段大小可提示突变位点的位置,切割产物的数量能够提示突变数量。在设计PCR 引物一般要求在500 bp 左右,目标位点不要设计在中央,酶切产生两条大小不同的条带。Surveyor 和T7EI 酶切的条带可以利用软件分析计算NHEJ 百分比。
电转染的Cas9和TALEN质粒对细胞基因组产生切割作用,则由于Surveyor和T7EI核酸酶对基因组扩增子错配位点的切割作用,会产生较小的条带;若没有对细胞基因组产生切割作用,则会有较大的、完整无缺的基因组扩增条带。阳性对照组只会产生较大的、完整无缺的基因组扩增条带(图 5)。
对于T7EI和Surveyor酶切条带不同的原因分析:对不同的不匹配类型有着不同的切割偏好,T7EI对HE删除和插入位点的偏好要比Survery强;而Survery超越T7EI的优越性是在异位替换和转换[12]。Survery能切割所有类型的不匹配位点,所以有些不是所要的切割位点,Survery也把它切开,另外,Surveyor还对酶切的中间产物有很强的偏好性,100 bp的条带属于酶切的中间产物经Surveyor再次切割产生,因此Surveyor酶切产生了5条带。
HRM的特点是高特异性和高灵敏度,检测灵敏度可以达到1%-0.1%,在大量样本、多个突变位点的筛查上,比传统的非均一性方法更方便、性价比更高[13, 14]。HRM可对任何扩增子上的未知突变进行筛查,不需要识别不同等位形式的引物或探针,不受突变碱基位点和种类的局限,既可以对未知突变进行筛查、扫描,又可以对已知突变进行分析。所以,相比定量探针法的突变分析和其他类型的快速突变分析法,应用面大大拓展,操作简便、快速,结果准确,成本也大大降低,成为近年来国外新兴的遗传学、方法学研究和应用热门。
HRM技术在PCR扩增后即可直接被用来对样品进行分析,不需要样品的分离纯化。因而可以降低分离纯化过程中所带来的样品污染的可能性,同时,也能大大减少工作量。HRM技术在PCR扩增时,一定要确保样品混匀,避免饱和染料混合不均,但是HRM技术对硬件的要求严格,对温度分辨率和仪器温度的均一性要求非常高,仪器需要高能量的激发光源以监测熔解曲线的微小变化,这种技术受限于仪器的使用。另外,HRM的引物最好在100-250 bp,扩增片段越小,检测特异性SNP的可能性越大,但是,小片段法也存在明显的缺陷,不能检测所有类型的SNP[15]。HRM技术与Surveyor nuclease 和T7EI相比的缺点是不能得知突变位点的位置也不能能计算NHEJ百分比。
4 结论实验采用了3种检测基因靶位点突变技术HRM、Surveyor nuclease和T7EI核酸酶,3种检测方法的结果都表明作用于绵羊MSTN基因第一外显子和第三外显子的CRISPR/Cas9和TALEN人工核酸酶的生物活性有明显的作用。
| [1] | Huang MC, Cheong WC, Lim LS, et al. A simple, high sensitivity mutation screening using Ampligase mediated T7 endonuclease I and Surveyor nuclease with microfluidic capillary electrophoresis[J]. Electrophoresis, 2012, 33(5):788-796. |
| [2] | Qiu P, Shandilya H, D'Alessio JM, et al. Mutation detection using Surveyor nuclease[J]. Biotechniques, 2004, 36(4):702-707. |
| [3] | Kim HJ, Lee HJ, Kim H, et al. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly[J]. Genome Res, 2009, 19(7):1279-1288. |
| [4] | Zhou L, Wang L, Palais R. High-resolution DNA melting analysis for simultaneous mutation scanning and genotyping in solution. Clin Chem, 2005, 51(10):1770-1777. |
| [5] | Cui G, Zhang L, Xu Y, et al . Development of ahigh resolution melt-ing Development of a high resolution melting method for genotyping of risk HLA-DQA1 and PLA2R1 alleles and ethnicdistribution of these risk alleles[J]. Gene, 2013, 514(2):125-130. |
| [6] | Er TK, Kan TM, Su YF, et al. High-resolution melting(HRM) analysis as a feasible method for detecting spinal muscular atrophy via dried bloodspots. [J]Clin Chim Acta, 2012, 413(21-22):1781-1785. |
| [7] | Lin Y, Cradick TJ, Bao G. Designing and testing the activities of TAL effector nucleases[J]. Methods Mol Biol, 2014, 1114:203-219. |
| [8] | Tricarico R, Crucianelli F, Alvau A. High resolution melting analysis for a rapid identification of heterozygous and homozygous sequence changes in the MUTYH gene[J]. BMC Cancer, 2011, 11:305. |
| [9] | Wittwer CT, Reed GH, Gundry CN. High-resolution genotyping by amplicon melting analysis using LCGreen[J]. Clin Chem, 2003, 49(6 Pt 1):853-860. |
| [10] | Zhou Y, Zhu S, Cai C, et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells[J]. Nature, 2014, 509(7501):487-491. |
| [11] | Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells[J]. Science, 2014, 343(6166):84-87. |
| [12] | Tsuji T, Niida Y. Development of a simple and highly sensitive mutation screening system by enzyme mismatch cleavage with optimized conditions for standard laboratories[J]. Electrophoresis, 2008, 29(7):1473-1483. |
| [13] | Reed GH, Wittwer CT. Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis[J]. Clin Chem, 2004, 50(10):1748-1754. |
| [14] | Liew M, Pryor R, Palais R. Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons[J]. Clin Chem, 2004, 50(7):1156-1164. |
| [15] | 张建佚, 冯洁, 杨泽, 等. 高分辨率熔解曲线小扩增子法结合混样法在线粒体13928G > C突变分析中的应用[J]. 中国医药生物技术, 2009, 6:445-448. |