




天然多胺包括腐胺、亚精胺和精胺,是广泛存在于真核细胞的一类低分子脂肪族阳离子化合物,是细胞生长的必需组分[1]。研究表明,多胺在DNA复制、细胞增殖、凋亡和动物繁殖过程中发挥重要作用[2]。正常生理条件下,细胞内多胺水平受到多胺生物合成酶、分解代谢酶及其跨膜转运系统的精确调控[3, 4]。大量研究表明,多胺与多胺合成酶在肿瘤细胞中含量异常升高,而且机体多胺稳态的紊乱与癌症的发生发展密切相关[5, 6]。因此,可通过调节多胺合成酶的活性和利用多胺跨膜转运系统来靶向治疗癌症。鸟氨酸脱羧酶(ornithine decarboxylase,ODC)和S-腺苷甲硫氨酸脱羧酶(S-adenosylmethionine decarboxylase,AdoMetDC)作为多胺生物合成的限速酶,对细胞内多胺水平的调节有着十分重要的作用。而多胺转运系统对多胺结构具有高度的特异性,能有效地将含有多胺结构的多胺类似物或多胺与药物缀合成的多胺缀合物转入细胞内。本文综述了近年来基于多胺代谢途径的相关抗癌治疗,以及通过多胺转运系统利用多胺类似物和多胺缀合物等药物进行抗癌治疗的研究进展,旨在为今后利用多胺代谢途径靶向治疗癌症的相关研究提供参考。
1 鸟氨酸脱羧酶在癌症治疗中的作用和机制多胺不仅能调控细胞增殖,而且与肿瘤的发生和转移密切相关。研究表明,在结肠癌和皮肤癌等上皮组织相关癌症的发生过程中,多胺水平和ODC表达显著增加[7]。且有研究表明,多胺代谢途径是Myc和Ras致癌基因的下游靶点,抑制多胺合成将扰乱这些基因的功能[8, 9, 10, 11]。ODC不仅是多胺合成途径中的第一限速酶,而且是致癌基因Myc的直接作用靶点[12]。因此,细胞内ODC的表达水平与癌症发生密切相关。Kubota等和Smith等[13, 14]研究发现过表达ODC不仅可以诱导细胞的恶性转化,还能增强细胞的侵袭性。2-氟甲基鸟氨酸(2-difluoromethylornithine,DFMO)可通过特异性抑制多胺合成途径的第一限速酶ODC,从而降低细胞内多胺含量,进而发挥其抗癌效应。
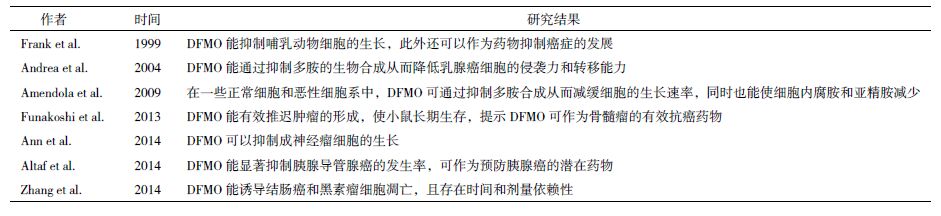
AdoMetDC是多胺合成代谢的第二限速酶[15],甲基乙二醛双脒腙(methylglyoxal bis-guanylhydrazone,MGBG)是AdoMetDC的竞争性抑制剂[16],但是其在调节多胺稳态过程中发挥的抗增殖效应可能与其线粒体毒性有关[17, 18]。SAM486A也是一种AdoMetDC竞争性抑制剂,其对线粒体的毒性损伤较小[19]。目前,SAM486A已经在多种癌症上进行了Ⅰ期和Ⅱ期临床试验[20]。在用SAM486A治疗非霍奇金淋巴瘤过程中,癌症缓解率可达19%。AdoDATO是亚精胺合成酶的特异性抑制剂,可以导致亚精胺减少,而腐胺和精胺含量仍然增加,不能很好的抑制癌细胞的生长[21]。另外,Holm和Pegg等[22]报道,一种类似于AdoDATO的精胺合成酶抑制剂也显示对抗癌治疗发挥的作用较小。总之,亚精胺和精胺合成酶抑制剂可能由于不能同时减少细胞内3种多胺的含量,从而限制了该类抑制剂的抗癌效应,然而其具体机制还需要进一步研究阐明。

|
| 图 1 靶向多胺代谢途径的抗癌治疗 |
细胞内多胺含量过高会导致癌症的发生,因此,干扰多胺代谢而耗竭细胞内的多胺,拮抗多胺的促细胞生长功能逐渐成为当今抗肿瘤治疗和药物设计的新策略[5, 23]。多胺类似物能与天然多胺竞争性结合多胺转运通道,借助多胺转运通道进入细胞内,下调多个多胺合成酶的活性,促进多胺的分解代谢,从而引起细胞内多胺的耗损,减少细胞内天然多胺的含量。因此,多胺类似物可作为潜在的抗肿瘤药物。此外,当天然多胺或人工合成多胺与细胞毒性药物缀合后,缀合物能被肿瘤细胞膜上的多胺转运体识别,并被运至细胞内,从而发挥靶向的抗肿瘤效应。因此,多胺类似物和多胺缀合物都可通过多胺转运体的介导进入细胞从而发挥其抗肿瘤效应。
3.1 减少机体多胺水平含有多胺基本结构的多胺类似物和缀合物能被多胺转运系统识别并被转运进入细胞内。因此,近年来研究者们尝试运用多胺结构作为载体将药物转运至肿瘤细胞造成细胞毒性进而达到抗癌治疗的目的。Delcros等[24]研究表明,一些多胺类似物能抑制多胺转运系统的活性,减少细胞对外源多胺的摄取,进而减少细胞内多胺水平。此外,Porter等[25]研究表明,多胺类似物BESPM不仅能抑制ODC和AdoMetDC的活性,而且能显著的增加黑素瘤细胞SAT的活性,促进多胺的降解,进而减少细胞内多胺水平。因此,多胺类似物不仅能抑制细胞对外源多胺的摄取,还能通过下调细胞多胺生物合成,增强细胞多胺的分解代谢,进而降低细胞内多胺水平,从而达到抗肿瘤效应。
3.2 调节组蛋白乙酰化水平多胺和多胺类似物能与DNA产生交互作用。在核心组蛋白赖氨酸的尾端修饰组蛋白被认为是调节基因表达的关键,而且这与癌症的发展和治疗密切相关[26]。组蛋白乙酰化状态异常将抑制肿瘤抑制基因的表达,因此组蛋白去乙酰化酶抑制剂在癌症治疗过程中发挥关键作用[27, 28]。研究表明,多胺能改变细胞内组蛋白乙酰转移酶和组蛋白去乙酰化酶抑制剂的活性[29, 30]。在小鼠皮肤过表达ODC,能显著增加细胞内多胺水平,并改变组蛋白乙酰转移酶和组蛋白去乙酰化酶抑制剂的活性[31]。而细胞内多胺浓度的变化所导致的核染色质的局部改变将增加某些特定原癌基因的表达,并抑制肿瘤抑制基因的表达[23]。因此,多胺类似物可以通过影响多胺代谢,靶向改变肿瘤细胞的乙酰化状态,进而在癌症治疗中发挥作用。
3.3 调节组蛋白甲基化水平组蛋白尾端的甲基化状态也在基因表达调控的过程中发挥重要作用。其中组蛋白末端的赖氨酸甲基化状态是决定甲基转移酶活性的关键。组蛋白赖氨酸特异性脱甲基酶1(Lysine-specific demethylase 1,LSD1)可通过与转录抑制因子作用从而沉默肿瘤抑制基因的表达[32]。在结肠癌细胞中,多胺类似物是LSD1的有效抑制因子,可恢复已沉默的肿瘤抑制基因的表达[33]。Zhu等[34]研究表明,在胸腺癌细胞中多胺类似物能有效的抑制LSD1,从而改变基因的表达和染色体结构。因此,多胺类似物能通过调节组蛋白的甲基化状态,从而发挥其抗肿瘤效应。
3.4 影响细胞周期和细胞凋亡研究表明,多胺类似物能诱导肿瘤细胞的程序性死亡。多胺类似物能诱导细胞内SAT和SMOX活性,在氧化分解细胞内多胺的同时产生H2O2,从而诱导细胞凋亡[35]。Tian等[36]研究表明,多胺类似物DENSPM处理胶质母细胞瘤细胞能诱导SAT高表达,从而能部分促进细胞脱离和凋亡。Stanic等[37]用DENSPM处理人C-28/12软骨细胞后,结果表明DENSPM能诱导细胞凋亡。王世召等[38]用DENSPM处理人胶质瘤LN299细胞后,其存活率随药物浓度的增加而逐渐降低,亚精胺/精胺-N1-乙酰基转移酶、多胺氧化酶、ODC水平上升,细胞内腐胺、亚精胺和精胺水平显著下降,提示DENSPM可能通过降低肿瘤细胞内多胺表达水平来抑制LN229细胞的生长并诱导其凋亡。因此,多胺类似物能通过诱导癌细胞凋亡从而发挥其抗癌效应。谢松强等[39]研究表明,多胺缀合物NNINspm可通过抑制p70S6K和mTOR蛋白的磷酸化,下调Bcl-2、CDK4,上调p27,从而使肝癌细胞HepG2细胞周期阻滞于G0/G1期,并诱导细胞凋亡。Yang等[40]研究表明,单萘酰亚胺-亚精胺缀合物(Mononaphthalimide-spermidine,MNISpd)可增强p21表达,减少cdc2表达,进而使HeLa细胞周期停滞;且MNISpd能激活caspase-3,抑制凋亡抑制蛋白XIAP的表达,从而诱导HeLa细胞凋亡。另外,Yang等[41]研究表明,MNISpd能通过诱导HeLa细胞细胞色素C的释放,提高caspase-3/9的活性,上调Bax蛋白和下调Bcl-2蛋白的表达,从而诱导细胞凋亡;进一步研究表明,MNISpd可通过原有的caspase依赖性途径和AIF介导的非caspase依赖途径诱导HeLa细胞凋亡。综上所述,多胺缀合物和多胺类似物可显著阻滞肿瘤细胞的细胞周期,诱导肿瘤细胞凋亡,作为抗肿瘤治疗药物有着十分广阔的前景。
4 结语上述研究表明,通过调控多胺代谢途径合成酶的活性,可有效缓解癌症的发展;利用多胺转运通道对多胺结构的高度特异性,将多胺类似物和缀合物转运进入细胞内,可通过减少细胞内多胺水平,调节组蛋白乙酰化和甲基化状态,诱导癌细胞凋亡等途径发挥抗癌效应。然而,目前多胺跨膜转运的具体机制仍不清楚。因此,多胺跨膜转运机制的研究将是未来多胺抗癌研究的热点,而针对多胺类似物和缀合物的相关研究将为开发抗癌药物提供新的切入点。
| [1] | Dai Z, Wu Z, Wang J, et al. Analysis of polyamines in biological samples by HPLC involving pre-column derivatization with o-phtha-laldehyde and N-acetyl-L-cysteine[J]. Amino Acids, 2014, 46:1557-1564. |
| [2] | He H, Kang B, Jiang DM, et al. Molecular cloning and mRNA expression analysis of ornithine decarboxylase antizyme 2 in ovarian follicles of the Sichuan white goose(Anser cygnoides)[J]. Gene, 2014, 545:247-252. |
| [3] | 王贵鸿, 马容, 康波, 等. 多胺跨膜物质转运的机制[J]. 动物营养学报, 2014, 26:3245-3250. |
| [4] | Kruczynski A, Vandenber I, Pillon A, et al. Preclinical activity of F14512, designed to target tumors expressing an active polyamine transport system[J]. Invest New Drugs, 2011, 29:9-21. |
| [5] | Gerner EW, Meyskens FL Jr. Polyamines and cancer:old molecules, new understanding[J]. Nat Rev Cancer, 2004, 4:781-792. |
| [6] | Thomas T, Thomas TJ. Polyamine metabolism and cancer[J]. Journal of Cellular and Molecular Medicine, 2003, 7:113-126. |
| [7] | 易星, 莫远亮, 姜冬梅, 等. 多胺的生物学功能及其调控机制[J]. 动物营养学报, 2014, 26:348-352. |
| [8] | Tobias KE, Shor J, Kahana C. c-Myc and Max transregulate the mouse ornithine decarboxylase promoter through interaction with two downstream CACGTG motifs[J]. Oncogene, 1995, 11:1721-1727. |
| [9] | Shantz LM, Levin VA. Regulation of ornithine decarboxylase during oncogenic transformation:mechanisms and therapeutic potential[J]. Amino Acids, 2007, 33:213-223. |
| [10] | Holtta E, Sistonen L, Alitalo K. The mechanisms of ornithine decarboxylase deregulation in c-Ha-ras oncogene-transformed NIH3T3 cells[J]. J Biol Chem, 1988, 263:4500-4507. |
| [11] | Ignatenko NA, Babbar N, Mehta D, et al. Suppression of polyamine catabolism by activated Ki-ras in human colon cancer cells[J]. Molecular Carcinogenesis, 2004, 39:91-102. |
| [12] | Funakoshi-tago M, Sumi K, Kasahara AT, et al. Critical roles of Myc-ODC axis in the cellular transformation induced by myeloproliferative neoplasm-associated JAK2 V617F mutant[J]. PLoS One, 2013, 8:e52844. |
| [13] | Kubota S, Kiyosawa H, Nomura Y, et al. Ornithine decarboxylase overexpression in mouse 10T1/2 fibroblasts:cellular transformation and invasion[J]. Journal of The National Cancer Institute, 1997, 89:567-571. |
| [14] | Smith MK, Goral MA, Wright JH, et al. Ornithine decarboxylase overexpression leads to increased epithelial tumor invasiveness[J]. Cancer Research, 1997, 57:2104-2108. |
| [15] | Pegg AE. S-Adenosylmethionine decarboxylase[J]. Essays Biochem, 2009, 46:25-45. |
| [16] | Williams-ashman HG, Schenone A. Methyl glyoxal bis(guanylhy-drazone)as a potent inhibitor of mammalian and yeast S-adenosy-lmethionine decarboxylases[J]. Biochemical and Biophysical Research Communications, 1972, 46:288-295. |
| [17] | Pleshkewych A, Kramer DL, Kelly E, et al. Independence of drug action on mitochondria and polyamines in L1210 leukemia cells treated with methylglyoxal-bis(guanylhydrazone)[J]. Cancer Research, 1980, 40:4533-4540. |
| [18] | Seiler N. Thirty years of polyamine-related approaches to cancer therapy. Retrospect and prospect. Part 2. Structural analogues and derivatives[J]. Current Drug Targets, 2003, 4:565-585. |
| [19] | Regenass U, Mett H, Stanek J, et al. CGP 48664, a new S-adenosylmethionine decarboxylase inhibitor with broad spectrum antiproliferative and antitumor activity[J]. Cancer Research, 1994, 54:3210-3217. |
| [20] | Nowotarski SL, Woster PM, Casero RA, JR. Polyamines and cancer:implications for chemotherapy and chemoprevention[J]. Expert Reviews in Molecular Medicine, 2013, 15:e3. |
| [21] | Pegg AE, Tang KC, Coward JK. Effects of S-adenosyl-1, 8-diamino-3-thiooctane on polyamine metabolism[J]. Biochemistry, 1982, 21:5082-5089. |
| [22] | Holm I, Persson L, Pegg AE, et al. Effects of S-adenosyl-1, 8-diamino-3-thio-octane and S-methyl-5'-methylthioadenosine on polyamine synthesis in Ehrlich ascites-tumour cells[J]. The Biochemical Journal, 1989, 261:205-210. |
| [23] | Casero RA, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases[J]. Nature Reviews Drug Discovery, 2007, 6:373-390. |
| [24] | Delcros JG, Tomasi S, Duhieu S, et al. Effect of polyamine homolo-gation on the transport and biological properties of heterocyclic amidines[J]. J Med Chem, 2006, 49:232-245. |
| [25] | Porter CW, Pegg AE, Ganis B, et al. Combined regulation of ornithine and S-adenosylmethionine decarboxylases by spermine and the spermine analogue N1 N12-bis(ethyl)spermine[J]. The Biochemical Journal, 1990, 268:207-212. |
| [26] | Jenuwein T, Allis CD. Translating the histone code[J]. Science, 2001, 293:1074-1080. |
| [27] | Marks P, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer:causes and therapies[J]. Nat Rev Cancer, 2001, 3:194-202. |
| [28] | Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy:is transcription the primary target?[J]. Cancer Cell, 2003, 4:13-18. |
| [29] | Hobbs CA, Paul BA, Gilmour SK. Elevated levels of polyamines alter chromatin in murine skin and tumors without global changes in nucleosome acetylation[J]. Experimental Cell Research, 2003, 290:427-436. |
| [30] | Hobbs CA, Paul BA, Gilmour SK. Deregulation of polyamine biosynthesis alters intrinsic histone acetyltransferase and deacetylase activities in murine skin and tumors[J]. Cancer Research, 2002, 62:67-74. |
| [31] | Hobbs CA, Gilmour SK. High levels of intracellular polyamines promote histone acetyltransferase activity resulting in chromatin hyperacetylation[J]. J Cell Biochem, 2000, 77:345-360. |
| [32] | Shi YJ, Matson C, Lan F, et al. Regulation of LSD1 histone demethylase activity by its associated factors[J]. Molecular Cell, 2005, 19:857-864. |
| [33] | Huang Y, Gerrne E, Murray Stewart T, et al. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes[J]. Proc Natl Acad Sci USA, 2007, 104:8023-8028. |
| [34] | Zhu Q, Huang Y, Marton LJ, et al. Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1(LSD1)and altering chromatin structure in human breast cancer cells[J]. Amino Acids, 2012, 42:887-898. |
| [35] | Pledgie A, Huang Y, Hacker A, et al. Spermine oxidase SMO(PAOh1), Not N1-acetylpolyamine oxidase PAO, is the primary source of cytotoxic H2O2 in polyamine analogue-treated human breast cancer cell lines[J]. The Journal of Biological Chemistry, 2005, 280:39843-39851. |
| [36] | Tian Y, Wang S, Wang B, et al. Overexpression of SSAT by DENSPM treatment induces cell detachment and apoptosis in glioblastoma[J]. Oncology Reports, 2012, 27:1227-1232. |
| [37] | Stanic I, Facchini A, Borzi RM, et al. The polyamine analogue N1, N11-diethylnorspermine can induce chondrocyte apoptosis independently of its ability to alter metabolism and levels of natural polyamines[J]. J Cell Physiol, 2009, 219:109-116. |
| [38] | 王世召, 田野, 江荣才, 等. 多胺类似物DENSPM对人胶质瘤LN229细胞增殖的抑制作用[J]. 中华神经外科疾病研究杂志, 2013, 12:101-105. |
| [39] | 谢松强, 李骞, 张亚宏, 等. 萘酰亚胺-多胺缀合物NNINspm通过PI3K/Akt信号通路诱导肝癌细胞凋亡[J]. 中国药理学通报, 2010, 26:169-174. |
| [40] | Yang L, Li W, Tian Z, et al. Mononaphthalimide spermidine conjugate induces cell proliferation inhibition and apoptosis in HeLa cells[J]. Toxicology in Vitro, 2011, 25(4):882-889. |
| [41] | Yang L, Zhao J, Zhu Y, et al. Reactive oxygen species(ROS)accumulation induced by mononaphthalimide-spermidine leads to intrinsic and AIF-mediated apoptosis in HeLa cells[J]. Oncology Reports, 2011, 25:1099-1107. |