




2. 上海科技馆自然史研究中心,上海 200041;
3. 青海民族大学 青海省青藏高原植物化学重点实验室,西宁 810007
2. Natural History Research Center, Shanghai Science and Technology Museum, Shanghai 200041;
3. Key Laboratory for Tibet Plateau Phytochemistry of Qinghai Province, Qinghai University for Nationalities, Xining 810007
手参(Gymnadenia conopsea),系兰科(Orchi-daceae)手参属(Gymnadenia)多年生草本植物,别名手掌参和佛手参,其根部呈手掌状,入藏药,对神经衰弱、肺虚咳嗽等病症效果显著,在《妙音本草》和《四部医典》等著名药典中均有记录手参的药方。手参属共有约20个物种[1],手参作为该属的代表种,在中国地区主要分布于青藏高原的河滩、灌丛中,适宜的生长海拔为2 700-3 600 m,分布区以西藏、甘肃和青海等地为主[2]。手参的自然繁殖能力较低且本身数量稀少,近年来,由于其药用价值和食用价值的开发,导致过度采挖,数量进一步大幅减少[3]。
植物谱系地理学的研究对象主要是经历过冰期的植物的遗传特性,第四纪冰期是距今最近的一次,在这次冰期之后由于基因的渐渗和杂交,物种出现了区域性的扩张和分化[4]。青藏高原是世界海拔最高且地域最广的高原之一,其独特的气候条件和地理隔离使之成为研究植物谱系地理学的重要区域。青藏高原植物分化主要受到高原隆起、气候剧变因素的影响[5]。许多植物的谱系地理学研究揭示了其扩散的机制,例如,对杉叶藻叶绿体基因的谱系研究表明杉叶藻居群的分化时间恰巧与横断山脉的隆起有关[6];对普通野生稻与栽培稻的谱系分析表明了栽培稻由普通野生稻驯化而来,我国南方地区是水稻的起源中心[7];利用叶绿体基因psbA-trnH、rps16和trnL-trnF对大叶柴胡的谱系地理学研究发现,位于两地的大叶柴胡在花色等表型上存在明显差异,后经分歧时间估计发现两种群分离与末次盛冰期相关[8]。综上可知,植物遗传分化深受地理隔离和气候变化的影响,我国青藏高原地带由于地壳隆起、末次冰期和横断山脉的影响,是地理隔离较为严重、气候变化较为剧烈的区域之一,也是研究植物种群分化的最佳地域之一。
之前对手参遗传特性的研究多采用等位酶、SSR和ITS等分子标记技术,其目的往往是对手参进行更科学准确的分类[9]。Scacchi等[10]通过7个等位酶对意大利16个种群的手参进行遗传多样性研究,结果显示,根据手参生境的不同,可以将其分为干湿两组,并认为这是存在生殖隔离的两个相似种。Gustafsson等[11]对手参的简单重复序列(SSR)进行研究,通过对5个位点的检测发现,手参在种间的遗传分化低于种内的变异。Campbell等[1]采集了英国的17个种群样品,使用SSR标记检测种群内等位基因频率,并以此对手参进行了更加细致的亚种分类;Stark等[12]对生长于德国的手参种群也进行了ITS、SSR分析,在分类学上支持了Campbell的结果。已有的手参生物学研究多是在弥补形态学上分类的缺陷。
本研究利用叶绿体基因作为分子标记,对青藏高原上的濒危物种手参进行谱系地理学和遗传多样性研究,再利用分歧时间数据对该物种的分化历程进行讨论,旨在初步揭示青藏高原上手参的遗传结构分布格局及其成因。
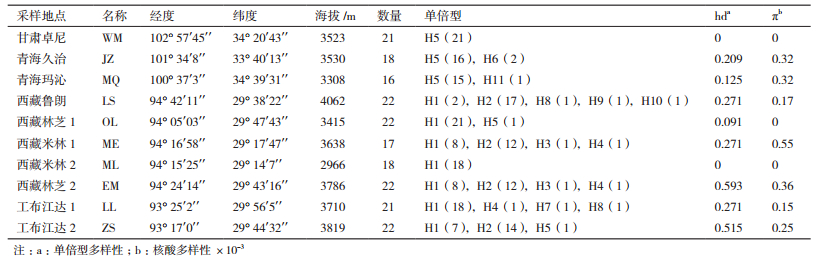
1 材料与方法 1.1 材料根据药典古籍记载和实地探索,本研究共采集到青藏高原上10个手参种群199个样本:其中西藏自治区7个种群144个样本,青海省2个种群34个样本,甘肃省1个种群21个样本。采样时每个样品之间的距离大于10 m,样品以手参叶为主,采样后放进装有硅胶的密封袋中保存。种群地理位置、海拔和数量等信息,见表 1和图 1。

|
| 图 1 手参种群11个单倍型的分布格局 |
本实验对4个叶绿体基因进行预实验,最后选取rbcl和psbA-trnH为最终片段,相应的引物分别为:rbcl(5’-ATGTCACCACAAACAGAG-AC-3’,5’-TCAAATTCAAACTTGATTTCTTTC-3’)[13];psbA-trnH(5’-GTTATGCATGAACGTAATGCTC-3’,5’-CGCGCATGGTGGATTCACAAATC-3’)[14]。
1.2.2 DNA提取每个干燥样品取50 mg,采用天根生化科技有限公司的试剂盒提取植物基因组DNA,试剂盒目录号为DP305。
1.2.3 扩增和测序使用筛选引物进行PCR扩增,退火温度设置为56℃,PCR扩增所用参数如下:95℃预变性4 min;94℃ 1 min,56℃ 1 min,72℃ 1 min,35个循环;72℃延伸7 min。对PCR产物进行1%浓度琼脂糖凝胶电泳检测,检测合格的样品送至上海美吉公司进行测序。
1.2.4 数据分析序列比对利用CLUSTAL W和MEGA 6完成[15, 16],使用软件DNASP 5计算单倍型多态性、核苷酸多态性和失配分析[17-19],利用软件HAPLONST计算种群内总遗传多样性hT、平均遗传多样性hS、种群遗传分化系数GST和NST;软件TCS 1.21被用来分析并绘制单倍型的演化网络图[20];系统发育分析和分歧时间估计由软件PAML完成[21];利用Arlequin软件对种群进行分组并做分子变异分析(AMOVA),同时计算群体遗传分化指数FST[22]。用软件RAxML和PAML软件包构建系统发生树和估计分歧时间,加入澳洲石斛(Dendrobium kingianum)、晶帽石斛(Dendrobium crystallinum)、绒叶斑叶兰(Goodyera velutina)和三蕊兰(Neuwiedia singapureana)作为外类群。
2 结果 2.1 样品采集和遗传多样性本研究共采集了青藏高原上的10个手参种群,以西藏、青海和甘肃三省为主,每个种群采集的样品数量在16-22之间,采样地点的海拔基本在3 000 m以上,各种群来源和地理位置等信息见表 1和图 1。
通过对10个种群199个样本的叶绿体DNA片段rbcl和psbA-trnH的PCR扩增和测序,得到rbcl片段的长度为1 280 bp,psbA-trnH的长度为663 bp,把两个基因分别比对、连接起来得到1 943 bp的叶绿体基因,组合起来的叶绿体基因中共检验得到12个变异位点,其中包括10个单核苷酸突变位点和2个插入缺失,序列变异位点见表 2。
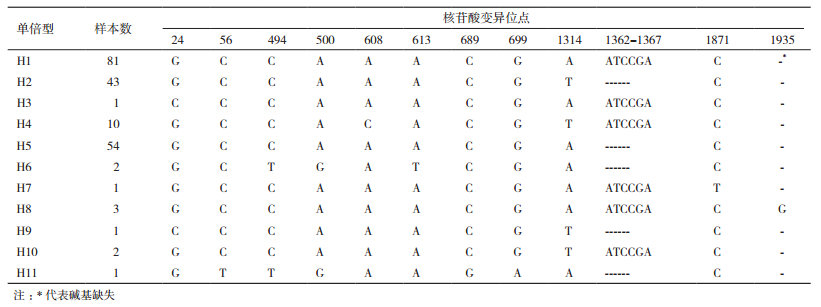
利用DNASP 5软件进行单倍型分析,共得到11个单倍型,分布情况见表 1和图 1。单倍型H1是西藏地区的主要单倍型,199个样品中共有81个属于单倍型H1;单倍型H2的数量仅次于H1,有43个样本;单倍型H5是青海甘肃地区的主要单倍型,也是西藏地区和青海甘肃地区唯一的共享单倍型。西藏鲁朗地区(LS)的单倍型最为丰富,有H1、H2、H8、H9和H10五种单倍型;很多种群都有其特有的单倍型,如:西藏林芝(EM)的H3、甘肃久治(JZ)的H6、西藏工布达江(LL)的H7、西藏鲁朗(LS)的H9、甘肃玛沁(MQ)的H11。
基于叶绿体基因的单倍型多样性(hd)在0-0.593之间,其中,甘肃卓尼(WM)和西藏米林(ML)的单倍型多样性值最小(hd=0),西藏林芝(EM)的单倍型多样性值最大(hd=0.593)。核苷酸多态性(π)在0-0.00 055之间,其中甘肃卓尼(WM)、西藏林芝(OL)和西藏米林(ML)的π值最小(0),西藏米林(ME)的π值最大(0.000 55),见表 1。
在TCS构图时,原始单倍型与后来衍生出来的单倍型往往呈现星状结构,原始单倍型处于星状结构的中心。在手参种群中,单倍型H1处于中心位置(图 2),可能是最原始的单倍型,在所有单倍型中,单倍型H1出现频率最高,尤其是在西藏片区的7个种群中,都有H1的存在。单倍型H5是青海甘肃地区的主要单倍型,从TCS网状图看,也是该地区较原始的单倍型。

|
| 图 2 手参种群11个单倍型的TCS网络图 |
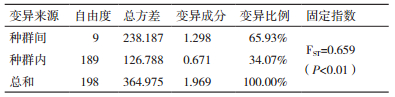
通过HAPLONST软件计算手参种群内遗传多样性HS值为0.285,总的遗传多样性HT值为0.762,种群间遗传分化系数GST和NST分别是0.626和0.398,NST小于GST,说明青藏高原上的手参种群没有明显的谱系地理结构,也就是亲缘关系较近的单倍型没有明显地出现在相同或者相近的种群中。
分子方差分析(AMOVA)的结果(表 3)表明,65.93%的分子变异来源于种群间,34.07%的变异来源于种群内部,固定指数FST的值为0.659(P < 0.01),种群间存在着显著的遗传分化。
用DNASP 5软件对手参的叶绿体DNA连接序列进行失配分析,结果(图 3)表明,青藏高原手参的失配分析曲线呈现多峰的形式,说明手参种群相对稳定,近期并没有出现突然扩张;同时,对手参种群进行中性检验,结果表明,Tajima’s D值为-1.038(P > 0.1),Fu and Li’s D值为-0.862(P > 0.1),不能拒绝中性选择零假设,支持失配分析的结果,即种群呈现稳定的分布状态。

|
| 图 3 手参种群的失配分析曲线 |
对叶绿体基因构建最大似然法(ML)的系统发生树,并进行分歧时间估计,结果(图 4)显示,单倍型在19.08 Mya开始分化,这一时期处于第三纪中新世时期(5.3 Mya-23 Mya),中新世为适度冰期气候,动植物已经相当程度地现代化,是喜马拉雅山和青藏高原隆起和环境变化的关键时期。多数单倍型分化时间在第四纪冰期之前的上新世,这一时期气候变得更加适宜生物发展,很多物种开始大量扩散生长。

|
| 图 4 手参种群单倍型的系统发育关系和分歧时间估计 注:*:节点数字的单位是百万年前(Mya) |
样品采集时,青海甘肃地区的3个种群与西藏地区的7个种群地理距离较远,但是在两个地区中间地带没有发现手参,而是采集到了与手参具有相似藏药功效的兰科植物掌裂兰(Dactylorhiza viridis),原因可能是过度采挖导致手参数量锐减或部分生境消失,或者藏药药典记载中把手参与掌裂兰(Dactylorhiza viridis)混淆。
在基于叶绿体基因的11个单倍型中,西藏地区拥有9个单倍型,数量多于青海和甘肃地区的3个单倍型,其中单倍型H5是西藏、青海和甘肃地区共有的单倍型,可能是连接青海甘肃地区和西藏地区的纽带。结合TCS网络图和系统进化分析,单倍型H1和单倍型H5有着较近的亲缘关系,其分歧时间在8.86百万年前(Mya)的中新世,这一时期喜马拉雅山的隆起导致亚洲季风模式发生改变,同时北半球的冰川作用也受影响,气候变化的压力可能使得单倍型H1经过多次变异形成了单倍型H5;然后经过风力或者鸟类等媒介作用,单倍型H5由西藏地区传播到青海甘肃地区,之后由于地理隔离和遗传漂变作用,单倍型H5演化出青海甘肃地区特有的单倍型H6和H11。
遗传分化系数GST只考虑单倍型频率,而NST还考虑了单倍型频率之间的遗传差异,一般认为NST显著大于GST时,种群间存在明显的谱系地理结构,否则不存在[23]。本研究中NST小于GST,说明青藏高原的种群间不存在明显的谱系地理结构。青藏高原上手参65.93%的分子变异来源于种群间,种群间分子差异显著大于种群内,固定指数FST为0.659(P < 0.01),基因流Nm值为0.17,表明种群间存在显著的遗传分化,种群间的基因交流比较弱,原因可能是青藏高原地区地势起伏、海拔落差很大,再加之手参种子主要是借助风媒传播,地理的阻隔限制了种群间的基因交流。
青藏高原是世界气候变化的敏感区,曾发生过快速隆起和强烈的第四纪冰川作用。关于青藏高原植物是如何应对冰期和演化的,目前主要有两种不同的模式观点:一是高原台面上的植物在冰期到来时灭绝了,现在的高原植物是冰期后从高原周边地区回迁到高原台面形成的;二是现代的高原植物起源于本地,在经历高原隆起和气候变化之后分化形成的,冰期到来时,植物种群生境破碎化形成多个避难所,冰期后逐渐恢复形成了现有分布格局[24]。
本研究中的失配分析和中性检验表明,手参在青藏高原上近期没有经历突然的扩张,种群保持相对稳定;系统发育分析和单倍型分歧时间估计显示,手参单倍型在12.48 Mya左右开始分化,这一时期正处于第三纪中新世时期(5.3-23 Mya),是青藏高原快速隆起的关键时期,由于青藏高原的隆起及其导致的气候变化,使得手参种群之间的基因流水平较低,长期的遗传漂变作用导致种群间存在着明显的遗传分化,因此手参单倍型的分化可能与青藏高原的快速隆起关系密切。分歧时间估计结果还表明大多数单倍型形成于第四纪冰期(0.01-3 Mya)之前(图 4),青藏高原手参现代的遗传谱系分布格局在冰期到来前已经基本形成,因此我们猜测,在冰期到来时,手参种群的生境可能已经发生破碎化,在冰期过后逐渐恢复。总之,关于青藏高原上手参的形成和演化,本研究结果支持第二种模式,即青藏高原上的手参属于本地起源。
青藏高原极端的气候条件和特殊的地理环境,导致手参种群间的遗传分化显著,种群间存在着不同的单倍型,由于西藏地区的单倍型多样性更加丰富并且包含青海和甘肃地区的主要单倍型,因此,在加强手参遗传资源保护时要着重考虑西藏地区的种群,即保护好西藏地区的手参种群就可以有效的保护手参的遗传多样性。
4 结论青藏高原植物手参的种群之间存在显著的遗传分化,种群分布相对稳定,高原台面的手参起源于本地,其分化与青藏高原的快速隆起关系密切,手参的遗传谱系分布格局在第四纪冰期之前就已经基本形成。
| [1] | Campbell VV, Rowe G, Beebee TJ, et al. Genetic differentiation amongst fragrant orchids (Gymnadenia conopsea sl) in the British Isles. Botanical Journal of the Linnean Society, 2007, 155 (3): 349–360. DOI:10.1111/boj.2007.155.issue-3 |
| [2] | 杨永昌. 藏药志[M]. 西宁: 青海人民出版社, 1991. |
| [3] | 金洪, 王俊杰. 手掌参的组织培养研究. 内蒙古农牧学院学报, 1995, 16(2): 74–77. |
| [4] | Liu JQ, Sun YS, Ge XJ, et al. Phylogeographic studies of plants in China: advances in the past and directions in the future. Journal of Systematics and Evolution, 2012, 50 (4): 267–275. DOI:10.1111/j.1759-6831.2012.00214.x |
| [5] | Wen J, Zhang JQ, Nie ZL, et al. Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Front Genet, 2014, 5 (4): 1–16. |
| [6] | Chen JM, Du ZY, Sun SS, et al. Chloroplast DNA phylogeography reveals repeated range expansion in a widespread aquatic herb Hippuris vulgaris in the Qinghai-Tibetan Plateau and adjacent areas. PLoS One, 2013, 8 (4): 60948–60948. DOI:10.1371/journal.pone.0060948 |
| [7] | Wei X, Wang R, Cao L, et al. Origin of Oryza sativa in China inferred by nucleotide polymorphisms of organelle DNA. PLoS One, 2012, 7 (11): 49546–49546. DOI:10.1371/journal.pone.0049546 |
| [8] | Zhao C, Wang CB, Ma XG, et al. Phylogeographic analysis of a temperate-deciduous forest restricted plant (Bupleurum longiradiatum Turcz.) reveals two refuge areas in China with subsequent refugial isolation promoting speciation. Molecular Phylogenetics and Evolution, 2013, 68 (3): 628–643. DOI:10.1016/j.ympev.2013.04.007 |
| [9] | Gustafsson S, Lonn M. Genetic differentiation and habitat preference of flowering-time variants within Gymnadenia conopsea. Heredity, 2003, 91 (3): 284–292. DOI:10.1038/sj.hdy.6800334 |
| [10] | Scacchi R, De Angelis G. Isoenzyme polymorphisms in Gymnaedenia conopsea and its inferences for systematics within this species. Biochemical Systematics and Ecology, 1989, 17 (1): 25–33. DOI:10.1016/0305-1978(89)90038-0 |
| [11] | Gustafsson S, Thoren PA. Microsatellite loci in Gymnadenia conopsea, the fragrant orchid. Molecular Ecology Notes, 2001, 1 (1-2): 81–82. DOI:10.1046/j.1471-8278.2001.00033.x |
| [12] | Stark C, Michalski SG, Babik W, et al. Strong genetic differentiation between Gymnadenia conopsea and G. densiflora despite morphological similarity. Plant Systematics and Evolution, 2011, 293 (1-4): 213–226. DOI:10.1007/s00606-011-0439-x |
| [13] | Little DP, Barrington DS. Major evolutionary events in the origin and diversification of the fern genus Polystichum (Dryopteridac-eae). American Journal of Botany, 2003, 90 (3): 508–514. DOI:10.3732/ajb.90.3.508 |
| [14] | Taberlet P, Gielly L, Pautou G, et al. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Molecular Biology, 1991, 17 (5): 1105–1109. DOI:10.1007/BF00037152 |
| [15] | Thompson JD, Gibson TJ, Plewniak F, et al. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research, 1997, 25 (24): 4876–4882. DOI:10.1093/nar/25.24.4876 |
| [16] | Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 2013, 30 (12): 2725–2729. DOI:10.1093/molbev/mst197 |
| [17] | Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. Journal of Molecular Evolution, 1983, 19 (2): 153–170. DOI:10.1007/BF02300753 |
| [18] | Nei M. Molecular evolutionary genetics[M]. New York: Colum-bia University Press, 1987. |
| [19] | Rozas J, Sanchez-Delbarrio JC, Messeguer X, et al. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics, 2003, 19 (18): 2496–2497. DOI:10.1093/bioinformatics/btg359 |
| [20] | Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Molecular Ecology, 2000, 9 (10): 1657–1659. DOI:10.1046/j.1365-294x.2000.01020.x |
| [21] | Yang ZH. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution, 2007, 24 (8): 1586–1591. DOI:10.1093/molbev/msm088 |
| [22] | Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 2010, 10 (3): 564–567. DOI:10.1111/men.2010.10.issue-3 |
| [23] | Pons O, Petit R. Measwring and testing genetic differentiation with ordered versus unordered alleles. Genetics, 1996, 144 (3): 1237–1245. |
| [24] | 陈伟烈. 西藏植被[M]. 北京: 科学出版社, 1988. |