




在真核生物的系统发生研究中,由于线粒体、叶绿体等细胞器基因较易获得而被广泛使用,例如,Zhu等[1, 2]利用线粒体基因matR研究蔷薇科的系统发育关系。由于线粒体基因组在不同植物类群中差异较大(300-600 kb),并且部分寄生类群植物的线粒体基因组中存在核基因的插入[3, 4],这些特点在一定程度上限制了线粒体基因在植物系统发生研究中的应用。不同物种的叶绿体基因具有组成和排列相似、大都是直系同源基因且基因序列比较保守等特点[5],同时已有大量的植物叶绿体基因组信息被测序发表,这些因素使得叶绿体基因成为研究植物系统发生关系的常用材料之一[6-8]。然而,由于叶绿体基因属于单系遗传,携带的进化信息有限,难以揭示大类群内部深层次的系统发生关系。随着高通量测序技术的发展,快速、准确及大规模获取植物的核基因序列成为可能,因此目前越来越多的研究人员开始采用属于双亲遗传、携带更多遗传信息的核基因来研究植物的亲缘关系[9, 10]。近年来大量有关工作通过转录组测序手段获取被子植物或陆地植物的编码序列,在多种尺度开展植物的系统发生关系研究[11-13]。
与细胞器基因相比,核基因具有复杂的进化模式,包括基因重复和基因丢失。研究表明,被子植物在进化过程中经历过多次全基因组重复事件(Whole genome duplication,WGD)[14];十字花科祖先经历过α和β两次全基因组重复事件[15-17];还存在一定数量的种、属特异的全基因组重复事件。例如,白菜近期经历过独立的基因组三倍化[18],大豆也被证实近期发生过一次全基因组重复事件。祖先基因组多倍化事件导致的全基因组重复,可能在后代类群中存在丢失不同基因拷贝的现象[19],使得部分基因退回到单拷贝状态,这可能导致不同物种间保留的基因为旁系同源关系。由于旁系同源基因无法提供正确的物种分化信息,因此在一定程度上限制了核基因在植物亲缘关系研究中的应用。面对这一挑战,研究人员不断开发新的生物信息学方法,从大量物种的测序数据中获取可能具有直系同源关系的单拷贝或低拷贝基因来构建真实反映物种关系的进化树,并且已经取得了一定的进展[20, 21]。
十字花科(Brassicaceae)是一个公认的自然大科,全科分为338个属,共有3 700多个种[22],主要分布于地中海及西北美等北温带地区,同时也广泛分布于我国西南、西北及东北的高山区及丘陵区[23]。十字花科中不仅存在白菜、油菜等具有重要应用价值的作物,还包含多种适应干旱、盐碱、低温等不同极端环境的植物,被广泛用于分子生物学研究的模式植物拟南芥也属于这一类群,因此该科具有重要的经济和研究价值。目前已经公布了十字花科中19个物种的基因组测序信息[2, 24-33]。十字花科祖先以及物种分化以后等不同演化阶段发生过多次基因组多倍化并可能导致一定程度的物种辐射[34-37],使得十字花科具有丰富的物种多样性,为植物系统发生有关研究提供了优良的素材。
近年来发表的关于十字花科植物族属的界定及科内系统演化关系方面的工作主要依靠叶绿体基因和少量核基因提供的进化信息。Beilstein等[38]利用来自113个十字花科物种的叶绿体基因ndhF序列信息,将十字花科划分为主要的3个类群(Ⅰ、Ⅱ、Ⅲ);同时Franzke等[39]定义了扩展的类群Ⅱ(Ext-ended Ⅱ,EⅡ),该类群包含了原类群Ⅱ以及部分类群Ⅱ的并系类群中的物种。由于类群EⅡ的物种亲缘关系还存在争议,因此在十字花科进化树上EⅡ的内部关系经常以梳状结构呈现。最近Huang等[10]利用113个核基因将十字花科分为6个主要类群,在得到与前人研究一致的3个主要类群的基础上,同时较好地解决了类群EⅡ的系统发育关系。
在十字花科以及其它被子植物系统发生关系的研究中,少数基因集合所包含的物种演化信息不能反映一致的物种亲缘关系。为了消除少数核基因的具体选择在十字花科系统发生关系分析中的影响,本研究通过使用系统基因组学方法,从34个物种的转录组及7个物种的基因组中筛选出大量低拷贝核基因,并使用组分矢量方法分析氨基酸序列片段中所包含的共同祖征、演征信息,以期获得稳定的十字花科物种系统发生关系,为深入研究十字花科以及其它类群物种的系统发生关系提供新的视角。
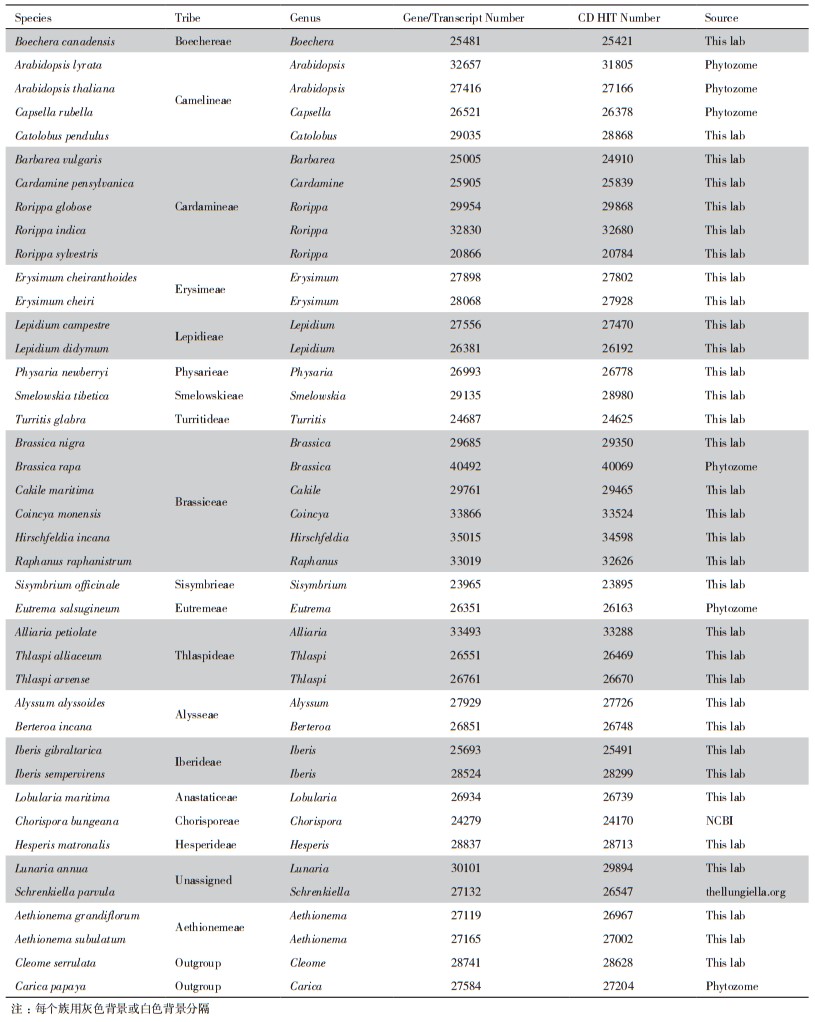
1 材料与方法 1.1 材料本研究采用了34个物种的转录组数据和7个物种的基因组数据,其中转录组数据包括来自于本实验室测序的33个物种的转录组信息以及公共数据库下载的1个物种的转录组信息(包括33个十字花科物种和1个醉蝶花科的Cleome serrulata作为外类群物种);基因组数据部分包括公共数据库下载的7个物种的基因组信息(包括6个十字花科物种和1个番木瓜科的番木瓜作为外类群物种),41个物种的具体信息及数据来源信息,见表 1。
利用Trinity软件对转录组测序数据进行拼接[40](参数为默认参数),使用TGICL软件(参数:-p 0.98,-l 40,-v)来获得更长的cDNA序列[41]。使用CD-HIT软件对基因组或转录组数据中相似度高的序列进行过滤处理[42]。最后进行序列比对及同源基因簇构建。为了准确识别41个物种的同源基因簇,我们对任意两物种间的蛋白质序列进行了all-against-all blastP比对分析,随后利用Inparanoid计算蛋白质序列全局比对相似性并舍去比对相似性低于40%的比对结果[43]。接下来利用OrthoMCL整合分析41个物种所包含的1 149 041个基因之间的蛋白质序列比对结果[44],最终得到35 948个同源基因簇,每个同源基因簇平均约包含29个基因,平均物种覆盖度为41.88%。
1.2.2 低拷贝基因的筛选标准以同源基因簇中单拷贝基因的物种数占总物种数的85%、80%及75%(对应35、33及31个具有单拷贝基因的物种数)作为阈值得到3个低拷贝同源基因簇集,作为后续分析的3个数据集。同时删除低拷贝同源基因簇中其它物种的多拷贝基因,保证每个低拷贝同源基因簇中所有物种均为单拷贝同源基因。
1.2.3 十字花科亲缘关系预测在每个数据集中分别提取每个物种的所有氨基酸序列组成该物种的“缩略基因组”。使用CVTree构建各个物种的组分矢量,同时计算两物种组分矢量间的夹角余弦值并将其转换为物种距离,最后使用邻接法(Neighbor Joining Method,NJ)构建十字花科物种系统发生树。
2 结果 2.1 物种基因组/转录组概况本研究选择来自于十字花科的39个物种(涵盖了18个族及两个未定族)和两个外类群物种。首先对其中34个物种的转录组数据进行了转录本拼接处理,得到了每个物种约25 000-35 000个转录本(表 1),每个转录本的平均氨基酸长度约为313(图 1)。由于高度相似的序列会对后续筛选低拷贝同源基因造成影响,因此对基因组数据以及拼接后的转录组数据中高度相似的序列进行了过滤处理。结果保留了平均每个物种约28 025个基因或转录本序列用于后续的相关分析。

|
| 图 1 41个物种的基因或转录本平均氨基酸长度分布 |
本研究对41个物种的氨基酸序列进行了all-against-all blastP序列比对分析,得到了两两物种间的同源基因对分布情况。随后,整合了所有物种的比对结果信息,进一步分析得到了41个物种间共计35 948个同源基因簇,其中平均每个同源基因簇中约含有来自17个物种的29个基因。通过观察以上这些同源基因簇中基因数目的分布情况,得到了两个较为集中分布的同源基因簇集,分别对应直方图上的两个峰值(图 3-A)。其中第一个峰值周围的同源基因簇中约含有0-15个基因,这部分同源基因簇集代表十字花科内部分支部分物种的祖先基因;第二个峰值周围的同源基因簇中约含有40-50个基因,这部分同源基因簇集则代表十字花科物种分化前的祖先基因,因此这部分同源基因簇保留了更全面的十字花科物种的遗传信息。

|
| 图 2 基因选择对物种树正确性的影响 A:当不同物种中丢失的拷贝不同时,基因树不能反映正确的物种关系。B:当不同物种中丢失的拷贝相同时,基因树才能反映正确的物种关系。红色星形代表基因重复事件,黑叉代表基因丢失事件。不同的物种用大写字母表示,基因用小写字母表示,重复基因的不同拷贝用数字表示 |

|
| 图 3 总同源基因簇(A)及3个同源基因簇子集(B)的基因数分布情况 OC(Ortholog Cluster)代表同源基因簇,下同 |
表 2展示了41个物种中包含以及未包含在同源基因簇中的基因数目,其中未在同源基因簇中的基因属于物种特异的基因,我们将这部分基因称为孤儿基因(Orphan gene)。我们发现白菜被包含在同源基因簇中的基因数目均多于其它物种,这与白菜发生过近期基因组多倍化事件相一致。由于外类群物种番木瓜及Cleome serrulata与十字花科其它物种的亲缘关系较远,且十字花科物种分化前发生过全基因组重复,因此两个外类群物种在同源基因簇中的基因数目显著小于其它物种。

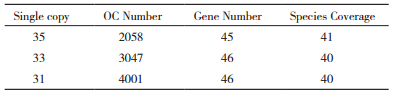
前期研究结果表明十字花科祖先在经历了两次全基因组重复后,其产生的复制基因迅速丢失。其中有约4 000对重复基因在十字花科物种分化前仍然保留了两个以上拷贝。由于重复基因在不同类群中可能保留了不同的拷贝,使得建树过程中可能采用了旁系同源基因而无法反映正确的物种关系(图 2)。为了尽量避免全基因组或基因重复事件对构建十字花科系统发育关系的影响,从同源基因簇中挑选满足一定物种覆盖度且各物种同源基因为单拷贝的基因集。以物种覆盖度为85%、80%及75%(对应35、33及31个物种数)作为阈值得到3个数据集合,分别包含2 058、3 047和4 001个单拷贝基因簇。在3种数据集中,大部分同源基因簇约包含40-50个基因(图 3-B),并且总物种覆盖度约为98%(表 3),表明3种数据集几乎覆盖所有物种,为后续的系统发生研究提供了较为充足的遗传信息。
利用CVTree方法及3种同源基因簇集合分别构建了十字花科系统发生树。随后整合分析了3种系统发育关系结果并最终得到了高度支持与稳定的十字花科系统发生树。如图 4所示,十字花科分为六大类群(类群A-F),与前人研究得到的十字花科六大类群的分类结果高度一致。其中类群A与类群B和C的结合类群互为姐妹类群,类群D为类群A、B、C结合类群的姐妹群,同时类群E为类群A、B、C、D结合类群的姐妹群,Aethionemeae族为基部类群F。在大尺度上,我们的结果与前人构建的十字花科三大类群(类群Ⅰ、Ⅱ、Ⅲ)的系统发育关系基本一致,即类群A和类群B分别对应类群Ⅰ和类群Ⅱ,同时类群Ⅲ中的大部分物种被划分至类群E中。在两个小类群C、D中,分别包含了类群EⅡ及类群Ⅲ中的部分物种。此外,我们还较好地解决了类群EⅡ的系统发育关系,将类群EⅡ中的大部分物种划分至类群B中(与类群Ⅱ互为姐妹类群),其它物种则被划分至类群C和类群D中。总体来说,基于CVTree方法得出的十字花科系统发育关系与前人利用叶绿体基因以及核基因构建的系统发育关系在大类群的亲缘关系上基本一致。在我们构建的系统发生树上(图 4),节点处的实心圆点代表该拓扑结构得到3种同源基因簇集合的共同支持,空心圆点代表该拓扑结构得到两种同源基因簇集合的共同支持。本研究系统发生树中几乎所有的节点都得到了高度支持,这也证明了该系统发育关系不随基因集合的变化而改变,因此具有较高的稳定性。

基于大量低拷贝核基因集合和CVTree方法构建的十字花科系统发生树在族、属等尺度上与前人的研究结果高度一致,在一些个别分支的系统发生关系上存在一定程度的差异。例如,本研究结果与Kagale等[12]的研究结果相一致,认为Lepidieae族的分化晚于Cardamineae族。而Huang等[10]的研究结果则认为Lepidieae族的分化时间较早,属于类群Ⅰ的基部分支。有研究表明Lepidieae族经历过异源多倍化事件[45],因此采用不同的同源基因集合可能会对正确反映Lepidieae族的分化位置造成影响。其次,在类群Ⅰ中Physaria newberryi的位置也与前人的研究结果存在差异,Huang等的研究结果中Physaria newberryi分支在c分支分化之后、b分支分化之前出现(图 4),而在我们的研究结果中Physaria newberryi位于类群Ⅰ的基部。我们发现Huang等[10]构建的系统发生树中,Physaria newberryi在类群Ⅰ物种中的枝长最长,表明该物种具有较快的进化速率,这可能会对不同的构树方法造成不同的影响。另外Huang等的研究认为Turritis glabra位于a类群基部(图 4),而在本结果中Turritis glabra与Boechera canadensis的亲缘关系更接近,并且为a类群的姐妹群,这可能与Huang等的结果中包含Alyssopsis mollis、Murbeckiella pinnatifida等Turritis glabra的近邻物种有关。
由于Schrenkiella parvula和Eutrema salsugineum均为耐盐植物,早期研究认为这两个物种同属于盐芥属(Thellungiella)。Kagale[12]和Huang等[10]的研究结果均认为Schrenkiella parvuls的分化早于Eutrema salsugineum和其它EⅡ-B类群物种,暗示这两个物种的抗盐性状为物种分化后单独获得。而本研究结果与早期的分类保持一致,即Schrenkiella parvula和Eutrema salsugineum的亲缘关系更接近,倾向于支持两个物种盐适应能力的获得发生在物种分化之前。此外,本研究结果稳定支持Lunaria annua与类群Ⅱ和EⅡ的结合类群互为姐妹类群,即该物种在d分支分化之后出现(图 4),而Huang等的研究结果显示Lunaria annua在d分支分化之前、e分支分化之后出现。由于d和e两个分支的进化关系较近,这也在一定程度上加大了正确反映Lunaria annua系统发生关系的难度,因此可能需要加入其它近邻物种才能确定Lunaria annua的进化位置。
基因重复和丢失等因素可能在一定程度上对依赖于直系同源基因比较的进化研究造成影响。在本研究结果中Brassiceae族物种约含有3万-4万个编码基因(表 1),显著高于十字花科其它族物种的平均基因数目。这与Brassiceae族祖先经历过近期基因组多倍化事件相符,表明Brassiceae族物种在全基因组重复后保留了大量的旁系同源基因。与前人的研究结果相比,本研究结果中Brassica nigra和Brassica rapa的位置发生了调换,这可能与Brassiceae族物种中普遍存在的旁系同源基因的保留、丢失模式有关,因此需要进一步深入研究才能为Brassiceae族构建出正确的系统发生关系。
综上所述,基于CVTree方法我们得到了稳定支持的十字花科物种系统发育关系。我们将十字花科划分为六大类群,其中类群A、B和E基本与前人研究结果中的类群Ⅰ、Ⅱ和Ⅲ一一对应,同时还增加了两个新类群(类群C、D)。此外,我们将EⅡ类群中大部分物种划分至类群B中,并且与类群Ⅱ互为姐妹类群,其它之前被认为属于类群EⅡ的物种则分布在类群C和类群D中。本研究结果在分类情况、各个类群内及类群间的系统发生关系上与前人的研究结果基本一致,同时较好地解决了类群EⅡ分类的争议。以上结果表明,在全基因组尺度上进行同源基因聚类以及低拷贝同源基因筛选,将在一定程度上减少使用少数基因构建进化树研究中面临的基因选择问题,使得快速准确地获得物种进化关系成为可能。快速发展的高通量测序技术使得CVTree方法不仅适用于原核生物、真菌等微生物的系统发生分析[46],也可以被应用在十字花科或更多类群物种的进化研究中。
4 结论本研究采用39个十字花科物种及两个外类群物种,利用系统基因组学方法筛选低拷贝核基因并基于组分矢量方法得到了高度支持与稳定的十字花科系统发育关系。本结果在分类情况、各个类群内及类群间的系统发生关系上与前人研究结果基本一致,同时较好地解决了类群EⅡ分类的争议,为使用低拷贝核基因深入分析十字花科系统发生关系提供了框架。
| [1] | Zhu XY, Chase MW, Qiu YL, et al. Mitochondrial matR sequences help to resolve deep phylogenetic relationships in rosids. BMC Evol Biol, 2007, 7 : 217. DOI:10.1186/1471-2148-7-217 |
| [2] | Anderson JT, Wagner MR, Rushworth CA, et al. The evolution of quantitative traits in complex environments. Heredity (Edinb), 2014, 112 (1): 4–12. DOI:10.1038/hdy.2013.33 |
| [3] | Bergthorsson U, Adams KL, Thomason B, et al. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature, 2003, 424 (6945): 197–201. DOI:10.1038/nature01743 |
| [4] | Westwood JH, Yoder JI, Timko MP, et al. The evolution of parasitism in plants. Trends Plant Sci, 2010, 15 (4): 227–235. DOI:10.1016/j.tplants.2010.01.004 |
| [5] | Olmstead R, Palmer J. Chloroplast DNA systematics: a review of methods and data analysis. American Journal of Botany (USA), 1994, 81 (9): 1205–1224. DOI:10.2307/2445483 |
| [6] | Moore MJ, Soltis PS, Bell CD, et al. Phylogenetic analysis of 83 plas-tid genes further resolves the early diversification of eudicots. Proc Natl Acad Sci USA, 2010, 107 : 4623–4628. DOI:10.1073/pnas.0907801107 |
| [7] | Jansen RK, Saski C, Lee SB, et al. Complete plastid genome sequences of three Rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol Biol Evol, 2011, 28 (1): 835–847. DOI:10.1093/molbev/msq261 |
| [8] | Weng ML, Ruhlman TA, Gibby M, et al. Phylogeny, rate variation, and genome size evolution of Pelargonium (Geraniaceae). Mol Phylogenet Evol, 2012, 64 (3): 654–670. DOI:10.1016/j.ympev.2012.05.026 |
| [9] | Zimmer EA, Wen J. Using nuclear gene data for plant phylogenetics: Progress and prospects Ⅱ. Next-gen approaches. Journal of Systematics and Evolution, 2015, 53 (5): 371–379. DOI:10.1111/jse.12174 |
| [10] | Huang CH, Sun R, Hu Y, et al. Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Molecular Biology and Evolution, 2016, 33 (2): 394–412. DOI:10.1093/molbev/msv226 |
| [11] | Zeng L, Zhang Q, Sun R, et al. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nature Communications, 2014, 5 : 4956. DOI:10.1038/ncomms5956 |
| [12] | Kagale S, Robinson SJ, Nixon J, et al. Polyploid evolution of the Brassicaceae during the Cenozoic era. Plant Cell, 2014, 26 (7): 2777–2791. DOI:10.1105/tpc.114.126391 |
| [13] | Yang Y, Moore MJ, Brockington SF, et al. Dissecting molecular evolution in the highly diverse plant clade Caryophyllales using transcriptome sequencing. Molecular Biology and Evolution, 2015, 32 (8): 2001–2014. DOI:10.1093/molbev/msv081 |
| [14] | Jiao Y, Wickett NJ, Ayyampalayam S, et al. Ancestral polyploidy in seed plants and angiosperms. Nature, 2011, 473 (7345): 97–100. DOI:10.1038/nature09916 |
| [15] | Bowers JE, Chapman BA, Rong J, et al. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature, 2003, 422 (6930): 433–438. DOI:10.1038/nature01521 |
| [16] | Barker MS, Vogel H, Schranz ME. Paleopolyploidy in the Brassicales: analyses of the Cleome transcriptome elucidate the history of genome duplications in Arabidopsis and other Brassicales. Genome Biol Evol, 2009, 1 : 391–399. |
| [17] | Tang H, Bowers JE, Wang X, et al. Synteny and collinearity in plant genomes. Science, 2008, 320 (5875): 486–488. DOI:10.1126/science.1153917 |
| [18] | Wang X, Wang H, Wang J, et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet, 2011, 43 (10): 1035–1039. DOI:10.1038/ng.919 |
| [19] | Xu G, Ma H, Nei M, et al. Evolution of F-box genes in plants: different modes of sequence divergence and their relationships with functional diversification. Proc Natl Acad Sci USA, 2009, 106 (3): 835–840. DOI:10.1073/pnas.0812043106 |
| [20] | Fulton TM, Van der Hoeven R, Eannetta NT, et al. Identification, analysis, and utilization of conserved ortholog set markers for comparative genomics in higher plants. Plant Cell, 2002, 14 (7): 1457–1467. DOI:10.1105/tpc.010479 |
| [21] | Wu F, Mueller LA, Crouzillat D, et al. Combining bioinformatics and phylogenetics to identify large sets of single-copy orthologous genes (COSⅡ) for comparative, evolutionary and systematic studies: a test case in the euasterid plant clade. Genetics, 2006, 174 (3): 1407–1420. DOI:10.1534/genetics.106.062455 |
| [22] | Warwick SI, Al-Shehbaz IA, Sauder CA. Phylogenetic position of Arabis arenicola and generic limits of Aphragmus and Eutrema (Brassicaceae) based on sequences of nuclear ribosomal DNA. Botany, 2006, 84 (2): 269–281. |
| [23] | Zhou TY, Lu LL, Yang G, et al. Brassicaceae (Cruciferae). Flora of China, 2001, 8 : 1–200. |
| [24] | Gong Q, Li P, Ma S, et al. Salinity stress adaptation competence in the extremophile Thellungiella halophila in comparison with its relative Arabidopsis thaliana. Plant J, 2005, 44 : 826–839. DOI:10.1111/tpj.2005.44.issue-5 |
| [25] | Amasino R. Floral induction and monocarpic versus polycarpic life histories. Genome Biol, 2009, 10 (7): 228. DOI:10.1186/gb-2009-10-7-228 |
| [26] | Dassanayake M, Oh DH, Haas JS, et al. The genome of the extremophile crucifer Thellungiella parvula. Nat Genet, 2011, 43 (9): 913–918. DOI:10.1038/ng.889 |
| [27] | Hu TT, Pattyn P, Bakker EG, et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet, 2011, 43 (5): 476–481. DOI:10.1038/ng.807 |
| [28] | Rushworth CA, Song BH, Lee CR, et al. Boechera, a model system for ecological genomics. Mol Ecol, 2011, 20 : 4843–4857. DOI:10.1111/mec.2011.20.issue-23 |
| [29] | Wu HJ, Zhang Z, Wang JY, et al. Insights into salt tolerance from the genome of Thellungiella salsuginea. Proc Natl Acad Sci USA, 2012, 109 (30): 12219–12224. DOI:10.1073/pnas.1209954109 |
| [30] | Slotte T, Hazzouri KM, Agren JA, et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat Genet, 2013, 45 (7): 831–835. DOI:10.1038/ng.2669 |
| [31] | Verbruggen N, Juraniec M, Baliardini C, et al. Tolerance to cadmium in plants: the special case of hyperaccumulators. Biometals, 2013, 26 (4): 633–638. DOI:10.1007/s10534-013-9659-6 |
| [32] | Halimaa P, Blande D, Aarts MG, et al. Comparative transcriptome analysis of the metal hyperaccumulator Noccaea caerulescens. Front Plant Sci, 2014, 5 : 213. |
| [33] | Vekemans X, Poux C, Goubet PM, et al. The evolution of selfing from outcrossing ancestors in Brassicaceae: what have we learned from variation at the S-locus?. J Evol Biol, 2014, 27 (7): 1372–1385. DOI:10.1111/jeb.12372 |
| [34] | Vision TJ, Brown DG, Tanksley SD. The origins of genomic duplications in Arabidopsis. Science, 2000, 290 (5499): 2114–2117. DOI:10.1126/science.290.5499.2114 |
| [35] | Simillion C, Vandepoele K, Van Montagu MC, et al. The hidden duplication past of Arabidopsis thaliana. Proc Natl Acad Sci USA, 2002, 99 (21): 13627–13632. DOI:10.1073/pnas.212522399 |
| [36] | Couvreur TL, Franzke A, Al-Shehbaz IA, et al. Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae). Mol Biol Evol, 2010, 27 (1): 55–71. DOI:10.1093/molbev/msp202 |
| [37] | Edger PP, Heidel-Fischer HM, Bekaert M, et al. The butterfly plant arms-race escalated by gene and genome duplications. Proc Natl Acad Sci USA, 2015, 112 (27): 8362–8366. DOI:10.1073/pnas.1503926112 |
| [38] | Beilstein MA, Al-Shehbaz IA, Kellogg EA. Brassicaceae phylogeny and trichome evolution. Am J Bot, 2006, 93 (4): 607–619. DOI:10.3732/ajb.93.4.607 |
| [39] | Franzke A, Lysak MA, Al-Shehbaz IA, et al. Cabbage family affairs: the evolutionary history of Brassicaceae. Trends Plant Sci, 2011, 16 (2): 108–116. DOI:10.1016/j.tplants.2010.11.005 |
| [40] | Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol, 2011, 29 (7): 644–652. DOI:10.1038/nbt.1883 |
| [41] | Pertea G, Huang X, Liang F, et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics, 2003, 19 (5): 651–652. DOI:10.1093/bioinformatics/btg034 |
| [42] | Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 2006, 22 (13): 1658–1659. DOI:10.1093/bioinformatics/btl158 |
| [43] | O'Brien KP, Remm M, Sonnhammer EL. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res, 2005, 33 (Database issue): 476–480. |
| [44] | Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res, 2003, 13 (9): 2178–2189. DOI:10.1101/gr.1224503 |
| [45] | Lee JY, Mummenhoff K, Bowman JL. Allopolyploidization and evolution of species with reduced floral structures in Lepidium L. (Brassicaceae). Proc Natl Acad Sci USA, 2002, 99 (26): 16835–16840. DOI:10.1073/pnas.242415399 |
| [46] | Qi J, Luo H, Hao B. CVTree: a phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res, 2004, 32 (Web Server issue): 45–47. |