




2. 深圳华大基因研究院,深圳 518083
2. Beijing Genomics Institute, Shenzhen 518083
基于单细胞测序技术(single-cell sequencing)的变异检测方法极大促进了我们对大脑、血液系统、免疫系统,及其组成这些系统的细胞之间异质性的认识。随着二代测序技术的发展,单细胞测序为理解不同细胞类型分化过程中的基因组稳定性提供了新视角。例如,Hou等[1]将多重置换扩增(multiple displacement amplification,MDA)和单细胞深度测序相结合应用到原发性血小板增多症(一种血癌)和肾透明细胞癌(一种肾癌)的肿瘤内部遗传特征研究中,解决了用组织样本测序时无法解决的肿瘤高异质性难题。McConnell等[2]以单个脑神经元细胞为研究对象,通过单细胞测序在细胞中发现了大段的DNA缺失或重复突变。尤其在植入前筛查或诊断领域,通过对人极体活检和单细胞测序能够准确检测胚胎染色体非整倍体,从而挑选正常胚胎进行植入[3]。
单核苷酸多态性(single-nucleotide polymorphi-sms,SNPs)和拷贝数变异(copy number variations,CNVs)是两种主要的遗传多态类型。SNP是指由单个核苷酸改变而引起的DNA序列的改变,造成包括人类在内的物种之间染色体基因组的多样性,基于单细胞检测SNP通常需要30X以上的测序深度[4],测序成本较高。CNV是指与参照基因组比较,1 kb-1 Mb的DNA片段的缺失、插入、重复、倒位和复杂多位点的变异[5]。CNV包含数千个基因、疾病位点、功能性因子和部分重复序列,多发生在富含重复序列的位置。例如,端粒、着丝粒和异染色质;作为遗传标记,与SNP具有较好的互补性。目前荧光原位杂交(fluorescence in situ hybridization,FISH)技术、比较基因组杂交(array CGH,aCGH)、单核苷酸多态性-微阵列比较基因组杂交技术(single nucleotide polymorphism array based comparative genomic hybridization,SNP-array CGH)和高通量测序技术在检测单细胞CNV方面均有应用。
因为高通量测序成本较高,如何降低检测成本成为迫切需要解决的问题。低深度测序方法检测染色体变异方法具有成本低、准确性高、通量大等优点。Zong等[6]发现每一百个基因组位点中挑出一个位点进行测序就足够检测胚胎非整倍体。本研究从5例细胞系中挑选出单个细胞分别用两种常用的商业试剂盒进行全基因组扩增,用Hiseq2000进行低深度测序,旨在研究低深度测序在检测单细胞CNV方面的可行性。
1 材料与方法 1.1 材料5例B淋巴细胞系购于Coriell研究所,具体细胞系信息可根据表 1在网址https://catalog.coriell.org/上查询;RPMI 1640细胞培养基(含2 mmol/L的L-谷氨酰胺)、D-PBS缓冲液、胰酶-EDTA、未灭活的小牛血清、Qubit® dsDNA Broad-Range Assay kit购自Life Technologies公司;单细胞全基因组扩增试剂盒分别购自Sigma公司的GenomePlex® Single Cell WGA Kit和Rubicon Genomics公司的PicoPLEX® WGA Kit;在深圳华大基因研究院完成文库构建和Hiseq2000平台测序。

本研究在倒置显微镜下完成单细胞挑取,利用口吸管技术从悬浮细胞中挑取状态理想、形状圆润的单细胞。首先,吸取1 μL细胞悬浮液,在D-PBS缓冲液进行梯度稀释至能观察到单细胞分散的状态,用口吸管挑取单个细胞,然后在一新D-PBS缓冲液中进行洗涤至少3遍。最后,将洗涤干净的单细胞转移至预先装有4 μL D-PBS缓冲液(或单细胞全基因组扩增试剂盒提供的细胞裂解液)的0.2 mL的PCR管中,短暂离心后放置于-20℃暂时保存(一周内)或-80℃长期保存(一周以上)。
注意转移单细胞后的口吸管,可以返回洗涤液中反复吹打,以确保单细胞没有残留。
1.2.2 全基因组扩增分别将单细胞取出解冻后,进行短暂离心。同时利用两种单细胞全基因组扩增试剂盒按各自的操作说明进行扩增。
扩增完成后,用Qubit® 2.0 Fluorometer核酸荧光分析仪进行PCR产物浓度检测,计算扩增产量。然后取1-2 μL PCR产物进行1%琼脂糖凝胶电泳检测,检测PCR产物片段的长度范围以及统计扩增成功率。
1.2.3 文库构建和上机测序本项研究的数据在Illumina Hiseq2000测序仪上获得,文库构建方法按照Hiseq2000的PCR Free PE index文库构建标准操作进行,主要过程如下:首先取500 ng的PCR产物进行超声打断,目标片段范围为150-450 bp,然后进行末端修复,加3' -dA末端,以及连接测序的带标签接头。最后用Qpcr对文库进行浓度定量,将测序文库混合成上机文库进行上机测序,本研究选择SE50+8 index的测序类型。
1.2.4 数据质控和过滤对于测序下机数据首先通过FastQCv0.11.2软件对样本的数据指标进行质控;对于质控合格的样本去掉低质量,测序接头污染和扩增接头污染的序列,随机抽取2 M(百万条序列)序列用于后续分析。
1.2.5 序列比对利用短序列比对软件BWA(Version0.7.7)[7]将过滤后的序列比对到人类参考基因组上(GRCh37,UCSC release hg19);BWA的参数设置为bwa aln -l 15 -t 12,比对结果以SAM文件输出;根据SAM文件[8]中的FLAG,POS和MAPQ信息挑出非重复且唯一比对的序列用于下游分析,并统计样本比对指标。
1.2.6 均匀性评估基于之前报道的方法将参考基因组划分为长度不同的窗口,根据比对上序列的坐标信息,统计落入每个窗口内的序列数,之后对序列数进行标准化和GC含量矫正[9],得到代表每个窗口的拷贝数值,用每条染色体的变异系数来评估基因组的均匀性大小。
1.2.7 CNV检测对于单个样本,以全基因组范围内划分好的窗口为检测单元,对检验窗口中所包含的测序序列进行统计分析,用标准化后的落入每个窗口的序列数比值代表该窗口的测序深度。对不同GC含量的窗口计算每个GC含量间隔内的测序深度,并对相应GC含量的窗口进行深度校正。用wald-wolfowitz游程检验对全基因组内的窗口进行统计检验,找出不同于群体的窗口,将这些显著性窗口进行合并,最终根据合并区域的长度,深度值和显著性大小确定拷贝数变异的具体坐标[9]。
2 结果 2.1 试剂盒扩增产量表 2表示在最终体积一致的情况下两种不同试剂盒的扩增产量;图 1表示两种不同商业扩增试剂盒扩增浓度比较;从中可以看出同一样本Sigma公司的试剂盒(以下简称Sigma)扩增浓度较高,但样本间差异较大,而Rubicon公司(以下简称Rubicon)的样本间扩增浓度波动较小,且耗时较短。
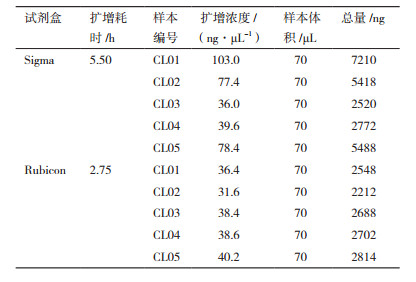

|
| 图 1 两种不同扩增试剂盒扩增浓度分布 |
表 3为经过数据过滤后单个样本比对的统计指标。Sigma试剂盒唯一比对率的波动范围为62.8%-65.1%,Rubicon试剂盒唯一比对率的波动范围为59.1%-60.1%。在唯一比对到参考基因组上的序列中Sigma扩增的重复序列占比为7.6%-20.1%,Rubicon的占比为2.5%-3.0%。另外Rubicon扩增的序列GC含量平均为43.5%,Sigma的平均为39.0%。

图 2为用变异系数衡量全基因组波动情况示意图。Sigma试剂盒的变异系数平均值±标准差为0.35±0.05,Rubicon试剂盒变异系数的平均值±标准差为0.28±0.04。使用SPSS软件对两组数据进行配对t检验,得出t=2.515,P=0.066。经过Rubicon处理的样本变异系数平均要比Sigma少0.07,说明Rubicon的均匀性较好。图 3显示样本各条染色体上标准化后的深度均在1附近波动,19号染色体GC含量是所有染色体中最高的,导致测到的序列较少。

|
| 图 2 两种试剂盒扩增样本的均匀性比较 |

|
| 图 3 样本内各个染色体上的深度分布 |
参考之前报道的检测CNV方法对10个样本数据进行检测,表 4中列出了最终的检测结果。可以看出检出的区带与标准区带均有80%以上的重合,根据之前报道[10]的判别标准可以认为1、3、4、5号细胞系两种试剂盒检出的区域与标准区域一致。在用Sigma扩增的产物中1个样本检出了8 Mb左右的假阳性;在用Rubicon扩增的产物中无假阳性信号产生。2号细胞系核型为46,XX,der(3)dup(3q)inv(p26q22)说明3号染色体在3q区带发生重复,且断裂和链接(倒位)发生于p26和q22区域,说明此数据分析方法不能检测倒位事件。从图 4可以看出采用低深度测序策略变异区域缺失信号明显,这也证明了低深度测序可以检出有效的CNV。


|
| 图 4 来自Sigma(左)和Rubicon(右)扩增样本的染色体变异检测结果 A:CL01_Sigma;B:CL01_Rubicon;C:CL02_Sigma;D:CL02_Rubicon;E:CL03_Sigma;F:CL03_Rubicon;G:CL04_Sigma;H:CL04_Rubicon;I:CL05_Sigma;J:CL05_Rubicon 红线为拟合后的区域,CopyRatio在0.7以下的为缺失,1.3以上的为重复 |
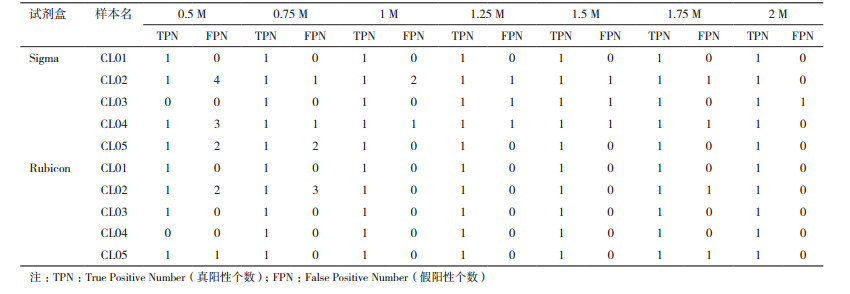
随机抽取0.5、0.75、1、1.25、1.5、1.75和2 M测序序列来评估鉴定拷贝数变异的最低数据量。从表 5中可以看出当数据量在0.75 M以上时可以有效检测出全部变异。
在人类已知的200多种染色体疾病中大多数是由染色体数目异常引起,如常见的13、18和21号染色体异常。另有部分染色体疾病因缺失或重复一段染色体片段而引起,统称为染色体微缺失/微重复综合征[11]。基于低深度测序对单细胞染色体非整倍体进行筛查在临床上已有报道[12],但缺乏基于此策略对染色体微小变异的报道。本实验对5例已知核型的单细胞采用低深度测序策略检测染色体微小变异,为这一策略在临床上的应用提供了数据支持。本实验发现Rubicon试剂盒具有较好的均匀性,原因可能在于Rubicon试剂盒先以原始基因组为模板进行复制,经过12次循环后,再以扩增产物为模板进行扩增可有效降低扩增误差,增加保真性[13]。而Sigma试剂盒是以扩增产物为模板进行扩增,其指数扩增会放大误差导致扩增均匀性差[14]。
最近,高分辨率寡核苷酸和单核苷酸多态性微阵列技术(single nucleotidepolymorphismarray,SNP array)也被用于产前诊断,与荧光原位杂交(fluore-scence in situ hybridization,FISH)、荧光定量聚合酶链式反应(polymerase chain reaction,PCR)和多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)相比,SNP array 能在更广范围内进行染色体异常的检测,可发现具有临床意义的、小于5 Mb 的染色体微缺失/微重复[15]。然而,对某些罕见的、数据库中未涵盖的CNV,仍无法实现可靠的诊断。随着二代测序技术在临床上的广泛应用,特别是单细胞检测领域,低深度的CNV筛查具有重要意义。例如,Navin等[16]利用单细胞测序定量基因组拷贝数变异数,通过对2例乳腺癌病人进行单细胞测序进而构建肿瘤演变模型,研究肿瘤细胞异质性;McConnell等[17]从3位死者体内分离了100个神经元,采用单细胞测序技术,结果显示41%的神经元拥有独特的CNV,表明这些神经元并不是来自于同一个亲本;在辅助生殖领域能够在D3对胚胎中的单个细胞进行染色体变异的精确筛查并在D5选择正常胚胎进行植入,使筛查和植入在同一个周期完成。低深度检测单细胞染色体微小变异还需更多的样本验证才能应用到临床领域,其将为分析遗传发育,疾病研究提供非常有价值的帮助。
4 结论本研究以来自相同细胞系的单细胞为材料,用两种不同的扩增试剂盒进行全基因组扩增,并进行低深度测序,结果表明低深度测序可以检测单细胞染色体的微小变异。
| [1] | Hou Y, Song L, Zhu P, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell, 2012, 148 (5): 873–885. DOI:10.1016/j.cell.2012.02.028 |
| [2] | McConnell MJ, Lindberg MR, Brennand KJ, et al. Mosaic copy num-ber variation in human neurons. Science, 2013, 342 (6158): 632–637. DOI:10.1126/science.1243472 |
| [3] | Van der Aa N, Zamani EM, Vermeesch JR, et al. Preimplantation genetic diagnosis guided by single-cell genomics. Genome Medicine, 2012, 5 (8): 71. |
| [4] | Ning L, Li Z, Wang G, et al. Quantitative assessment of single-cell whole genome amplification methods for detecting copy number variation using hippocampal neurons. Scientific Reports, 2014, 5 (5): 11415. |
| [5] | Goidts V, Cooper DN, Armengol L, et al. Complex patterns of copy number variation at sites of segmental duplications:an important category of structural variation in the human genome. Human Genetics, 2006, 120 (2): 270–284. DOI:10.1007/s00439-006-0217-y |
| [6] | Zong C, Lu S, Chapman AR, et al. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science, 2012, 338 (6114): 1622–1626. DOI:10.1126/science.1229164 |
| [7] | Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009, 25 (14): 1754–1760. DOI:10.1093/bioinformatics/btp324 |
| [8] | Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics, 2009, 25 (16): 2078–2079. DOI:10.1093/bioinformatics/btp352 |
| [9] | Zhang C, Zhang C, Chen S, et al. A single cell level based method for copy number variation analysis by low coverage massively parallel sequencing. PLoS One, 2013, 8 (1): e54236. DOI:10.1371/journal.pone.0054236 |
| [10] | Tsuang DW, Millard SP, Ely B, et al. The effect of algorithms on copy number variant detection. PLoS One, 2010, 5 (12): e14456. DOI:10.1371/journal.pone.0014456 |
| [11] | Gardner RJMK, Sutherland GR, Shaffer LG. Chromosome abnormalities and genetic counseling[M]. New York: Oxford University Press, 2004. |
| [12] | Wells D, Kaur K, Grifo J, et al. Clinical utilisation of a rapid low-pass whole genome sequencing technique for the diagnosis of aneuploidy in human embryos prior to implantation. Journal of Medical Genetics, 2014, 51 (8): 553–562. DOI:10.1136/jmedgenet-2014-102497 |
| [13] | Langmore JP. Rubicon Genomics, Inc. Pharmacogenomics, 2002, 3 (4): 557–560. DOI:10.1517/14622416.3.4.557 |
| [14] | Lei H, Fei M, Alec C, et al. Single-cell whole-genome amplification and sequencing:methodology and applications. Annu Rev, 2015, 6 : 10–14. |
| [15] | Schaaf CP, Wiszniewska J, Beaudet AL. Copy number and SNP arrays in clinical diagnostics. Annual Review of Genomics and Human Genetics, 2011, 12 (12): 25–51. |
| [16] | Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature, 2011, 472 (7341): 90–94. DOI:10.1038/nature09807 |
| [17] | McConnell MJ, Lindberg MR, Brennand KJ, et al. Mosaic copy number variation in human neurons. Science, 2013, 342 (6158): 632–637. DOI:10.1126/science.1243472 |