




2. 宁夏颅脑疾病重点实验室 国家重点实验室培育基地,银川 750004
2. Ningxia Key Lab of Cerebrocranial Diseases, the National Key Laboratory Incubation Base, Yinchuan 750004
自我复制是干细胞所特有的特性,在保持高度增殖能力的同时还能维持子代细胞处于不分化状态,并保持其多向分化潜能。肿瘤干细胞广泛存在于各种肿瘤中[1-6],也具有自我复制和多向分化的特性,并表达多种干细胞的标志分子,如CD34、CD133、ALDH1、CXCR4、Nestin、LRCs等。肿瘤干细胞一般处于休眠或者低增殖状态,但受到内、外因素刺激后,便可被活化进入自我复制和增殖状态,成为肿瘤发生的“种子”[7]。
组蛋白H3K9(dimethylated histone H3 lysine 9)甲基化修饰是发生在组蛋白H3的9位赖氨酸残基上的甲基化,与基因沉默相关[8]。在胚胎干细胞中,H3K9甲基化修饰的基因仅占基因组4%;当胚胎干细胞分化时,分化的细胞会失去H3K4与H3K27的双价可塑性修饰,转而以更为永久的DNA甲基化与H3K9甲基化为标记[9]。Sox2、c-myc、Oct4、nanog等是维持肿瘤干细胞自我复制至关重要的核心转录因子,在干细胞走向分化之后,这些基因必须被抑制,而这种抑制机制最可能与组蛋白H3K9甲基化修饰和DNA甲基化有关[10]。G9a是一种组蛋白赖氨酸(H3K9)甲基转移酶,催化常染色质H3K9的单甲基(H3K9me)和双甲基化(H3K9me2)[11, 12]。Bix01294是一种G9a抑制剂,可以促进胶质瘤细胞系的“神经球”克隆成球率,同时促进干细胞标志分子CD133和Sox2等干性基因的表达;转染G9a表达质粒则可以使CD133和Sox2表达水平明显下降[13]。
我们在前期工作[13]中发现,将U87细胞用加Bix01294的成球培养基培养5-7 d,可以得到质量较好的成球细胞,其CD133表达量明显升高,将此作为干细胞的模型;与此相对应,我们用贴壁培养的U87细胞转染G9a过表达质粒后2 d,这些细胞的CD133表达量有所下降,以此作为非干细胞的对照模型。在此两种干细胞和非干细胞的模型中,我们拟采用ChIP-seq(Chromatin immunoprecipitation-sequencing)技术对全基因组范围内H3K9me2修饰的基因进行分析比较,试图筛选出受H3K9me2修饰调控的“干性”相关基因。
1 材料与方法 1.1 材料U87细胞株(北京协和细胞库);DMEM培养基、胎牛血清、OPTI-MEM(HyClone公司);RNA提取试剂盒、脂质体试剂Lipofectamine 2000(INVITROGEN);B27添加剂(GIBCO);BFGF、EGF(INVITROGEN);Bix01294(Sigma公司);Q-PCR Mix(北京全式金);抗体均购自北京博奥森生物技术有限公司。
1.2 方法 1.2.1 细胞培养U87贴壁细胞用含10%胎牛血清、1%青/链霉素的DMEM培养基,置于37℃、5% CO2及饱和湿度的培养箱中培养。U87成球细胞用无血清培养基悬浮培养,培养基中加Bix01294(每1 mL培养基中加浓度为1 mmol/L的Bix 1 μL),5-7 d成球。无血清培养基(100 mL)配制:DMEM/F12:96 mL;B27 supplement(50×):2 mL;EGF(20 ng/ mL):4 µL;Basic-FGF(20 ng/ mL):40 µL;双抗:1 mL;谷氨酰氨:1 mL。
1.2.2 G9a表达质粒的构建及转染以人全基因组为模板,G9a引物进行PCR(引物由上海杰瑞生物公司代理合成)。用PCR产物、表达质粒pcDNA3.1构建G9a表达质粒,鉴定完全正确后进行转染。转染前一天,U87细胞接种于6 cm的培养皿中,无抗生素培养基贴壁培养,第2天细胞密度达到80%-90%,将21.2 μL的G9a表达质粒和20 μL Lipofectamine 2000混合液均匀滴入培养皿中,置培养箱6 h后换正常培养基。同样方法转染空载质粒。
1.2.3 Western blot检测G9a、H3K9me2和CD133蛋白的表达将转染空载质粒、G9a表达质粒3 d的贴壁细胞和悬浮培养的成球细胞分别提取总蛋白,BAC法测蛋白浓度,每泳道上样40 μg,10%SDS-PAGE胶电泳,PVDF膜湿转,5%脱脂牛奶封闭,一抗G9a(1:1 000)、H3K9me2(1:1 000)4℃冰箱孵育过夜,洗膜,二抗(1:1 000)室温孵育1 h,洗膜,滴加HRP发光液,曝光,显影。
1.2.4 ChIP-seq样本制备将贴壁和悬浮细胞分别离心,室温PBS重悬后加入终浓度为1%的甲醛轻微混匀,室温放置固定10 min;加入终浓度为0.125 mol/L的甘氨酸轻微混匀后置于冰上5 min,终止交联;1 500 r/min 4℃离心5 min,弃上清,用等体积的冷PBS洗涤细胞两次;1 500 r/min 4℃收集细胞,-80℃冻存。处理好的样本交于上海康成生物工程有限公司做ChIP-seq实验,公司做染色质免疫沉淀使用的抗体是H3K9me2抗体。样本在上机前进行质量检测,ChIP-seq样品要求DNA总量大于10 ng,片段主峰分布在100-500 bp之间。本实验样品:非干细胞片段主峰大小为234 bp,浓度为27.7 nmol/L;干细胞片段主峰大小为227 bp,浓度为15.3 nmol/L,符合上机测序要求,结果如图 1。

|
| 图 1 CHIP-seq样品质量分析 A:Bix01294成球细胞(干细胞);B:转染G9a表达质粒细胞(非干细胞) |
Trizol法分别提取干细胞和非干细胞的总RNA。以符合试验要求的RNA为模板逆转录cDNA,以SYBR Green为荧光标记物,GAPDH为内参,进行PCR扩增和荧光检测。反应体系:cDNA 2 μL,上游引物0.5 μL,下游引物0.5 μL,2×Master Mix 10 μL,ddH2O 7 μL。PCR扩增循环:95℃ 5 min,然后94℃ 5 s、60℃ 15 s、72℃ 10 s,经40次循环后72℃ 7 min。2-△△Ct法测定干细胞和非干细胞mRNA的相对表达量。QPCR引物由Premier Primer 5. 0 software设计。

Western blot结果(图 2)显示,U87细胞转染G9a表达质粒后,G9a和H3K9me2蛋白的表达量明显升高,说明G9a表达质粒转染成功,同时干细胞标志分子CD133和干性相关基因Sox2的表达量显著下降。加Bix01294的成球U87细胞,其G9a和H3K9me2蛋白的表达量显著下降,但CD133和Sox2的表达量却明显增加。

|
| 图 2 Western blot检测干细胞和非干细胞G9a、CD133、Sox2蛋白的表达情况 control:正常U87贴壁细胞;empty:转染空载质粒的U87贴壁细胞;G9a:转染G9a表达质粒的U87贴壁细胞(非干细胞);Bix01294:加Bix01294的U87成球细胞(干细胞) |
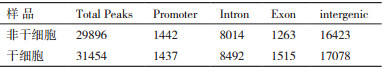
为了比较干细胞和非干细胞全基因组H3K9me2修饰的差异,分别统计了干细胞和非干细胞全基因组H3K9me2的富集情况(即peaks数)及全基因组上的启动子区、外显子区、内含子区、基因间区共4个区域上H3K9me2的富集情况(即peaks数)。结果(表 2)显示,在干细胞和非干细胞的全基因组上,H3K9me2修饰差异不明显,而且大多数H3K9me2 peaks分布在基因间区。
使用UCSC基因组浏览器,对胶质瘤干细胞和非干细胞的第6号染色体的H3K9me2分布情况进行可视化展示,结果如图 3所示,第6号染色体上干细胞中H3K9me2的富集情况稍小于非干细胞中H3K9me2的富集情况。

|
| 图 3 干细胞和非干细胞中H3K9me2修饰水平在6号染色体上的分布情况 |
ChIP-seq分别筛选干细胞和非干细胞中受H3K-9me2调控的基因,结果(图 4)显示在转录起始位点(Transcription Start Site,TSS)±2 000 bp范围内,胶质瘤干细胞中受H3K9me2调控的基因有1 006个,非干细胞中受H3K9me2调控的基因有976个,其中有355个同时存在于干细胞和非干细胞中。受H3K9me2调控的转录因子,胶质瘤干细胞中254个,非干细胞中238个,同时存在于干细胞和非干细胞中的95个。从转录因子中随机选取10个做QPCR检测,其中6个选自干细胞,4个选自非干细胞。所选取转录因子的名称及峰值位置见表 3。

|
| 图 4 干细胞与非干细胞进行ChIP-seq实验筛选得到的相关基因 |
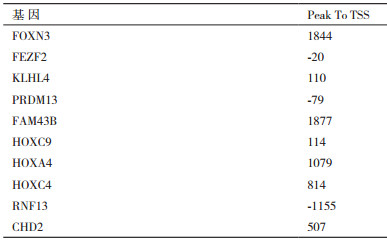
对随机选取的10个转录因子做QPCR验证,结果(图 5)显示干细胞中随机选取的FOXN3、FEZF2、KLHL4、PRDM13、FAM43B、HOXC9在干细胞中mRNA的相对表达量高于非干细胞,其中PRDM13、KLHL4的表达量增加显著,而从非干细胞中随机选取的HOXA4、HOXC4、RNF13、CHD2在干细胞中mRNA的相对表达量低于非干细胞,尤以CHD2表达量的降低最为明显。

|
| 图 5 干细胞和非干细胞中H3K9me2修饰的转录因子的mRNA相对表达量 |
ChIP-seq结果(图 6)显示,HOXC9在干细胞中H3K9me2修饰水平低于非干细胞,而CHD2在干细胞中H3K9me2修饰水平高于非干细胞,KLHL4变化不明显。与QPCR实验结果基本相符。

|
| 图 6 KLHL4、HOXC9、CHD2在干细胞和非干细胞中H3K9me2的修饰水平 |
GO结果(图 7)显示,胶质瘤干细胞基因主要参与的生物学过程有乳腺腺泡和腺管的发育、肢体的形成、参与重力反应、肌细胞融合、胚胎消化道的发生、环核苷酸分解代谢过程、光的传入、联会复合体的组装及合胞体的形成;主要参与的细胞组分有轴突的形成、神经肌肉接头的传递及肌肉的收缩、递质贮存释放、G蛋白等介导的细胞膜的信号转导、应激、钾通道;参与的分子功能主要有腺苷酸环化酶、MAP激酶、酪氨酸、丝氨酸、苏氨酸磷酸酶、二肽酶、环GMP磷酸二酯酶、钾通道和作用于碳-氮键水解酶的活性,以及肾上腺素受体、翻译起始因子、TBP类蛋白的结合。

|
| 图 7 胶质瘤干细胞基因的GO分析 |
不论是在肿瘤细胞还是普通细胞中,干性都不是一个稳定的状态[14, 15]。被普遍认为的肿瘤干细胞从静态变为动态的说法得到不断的证实,这种转变与肿瘤干细胞干性的得失密切相关[16]。在动态肿瘤干细胞理论中,已分化的肿瘤细胞通过去分化的方式,可重新获得干细胞特性[17]。肿瘤干细胞的特性主要依赖于特定基因的表达开放与关闭[18]。随着基因组测序技术的进步和表观遗传学迅速的发展,越来越多的研究者意识到,表观遗传学在肿瘤的调控中也占有一席之地[19]。表观遗传学包括DNA甲基化、组蛋白修饰、染色质重塑和RNA编辑等,它们在基因的核苷酸序列不发生改变的情况下调控基因的表达及细胞表型。如组蛋白甲基转移酶EZH2催化组蛋白形成H3K9me和H3K27me。研究表明,在T细胞淋巴瘤[20]、肝癌、结肠癌、前列腺癌、乳腺癌、卵巢癌等多种肿瘤中,上调EZH2的表达水平,肿瘤干细胞数量增多[21-25],而药物阻断或下调EZH2,则导致肿瘤干细胞的自我增殖能力减弱,致瘤能力抑制[26, 27]。
本实验主要通过ChIP-Seq方法筛选得到了一批峰值在TSS±2 000 bp范围内,而且在胶质瘤干细胞中高表达而在非干细胞中低表达的干性基因,例如FEZF2、KLHL4、PRDM13、HOXC9等;QPCR验证结果显示干细胞中随机选取的FOXN3、FEZF2、KLHL4、PRDM13、FAM43B、HOXC9在干细胞中mRNA的相对表达量高于非干细胞,尤其KLHL4、PRDM13的表达量升高明显,提示在干细胞中FOXN3、FEZF2、KLHL4、PRDM13、FAM43B、HOXC9的H3K9me2修饰水平低,被抑制的程度弱,因此在干细胞中的表达量增加。而非干细胞中随机选取的HOXA4、HOXC4、RNF13、CHD2在干细胞中的mRNA的相对表达量却低于非干细胞,提示在非干细胞中HOXA4、HOXC4、RNF13、CHD2的H3K9me2修饰水平高,被抑制的程度强,因此在干细胞中的表达量降低。此外,GO分析结果显示,胶质瘤干细胞基因主要参与的生物学过程有乳腺腺泡和腺管的发育、肢体的形成、参与重力反应、肌细胞融合、胚胎消化道的发生、环核苷酸分解代谢过程、光的传入、联会复合体的组装及合胞体的形成;主要参与的细胞组分有轴突的形成、神经肌肉接头的传递及肌肉的收缩、递质贮存释放、G蛋白等介导的细胞膜的信号转导、应激、钾通道;参与的分子功能主要有腺苷酸环化酶、MAP激酶、酪氨酸、丝氨酸、苏氨酸磷酸酶、二肽酶、环-GMP磷酸二酯酶、钾通道和作用于碳-氮键水解酶的活性,以及肾上腺素受体、翻译起始因子、TBP-类蛋白的结合。
4 结论本研究通过ChIP-seq技术比较了H3K9me2修饰在干细胞与非干细胞中的差异,筛选得到了一批H3K9me2修饰调控的“干性”相关基因:FOXN3、FEZF2、KLHL4、PRDM13、FAM43B、HOXC9等,并对TSS±2 000 bp范围内的基因进行了GO分析。此外,对随机选取10个转录因子进行QPCR验证,结果与ChIP-seq基本相符。
| [1] | Li YF, Xiao B, Tu SF, et al. Cultivation and identification of colon cancer stem cell-derived spheres from the Colo205 cell line. Braz J Med Biol Res, 2012, 45 (3): 197–204. DOI:10.1590/S0100-879X2012007500015 |
| [2] | Nakanishi Y, Seno H, Fukuoka A, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet, 2013, 45 (1): 98–103. |
| [3] | Chen J, Li YJ, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature, 2012, 488 (7412): 522–526. DOI:10.1038/nature11287 |
| [4] | Machado HL, Kittrell FS, Edwards D, et al. Separation by cell size enriches for mammary stem cell repopulation activity. Stem Cells Transl Med, 2013, 2 (3): 199–203. DOI:10.5966/sctm.2012-0121 |
| [5] | Driessens G, Beck B, Caauwe A, et al. Defining the mode of tumour growth by clonal analysis. Nature, 2012, 488 (7412): 527–530. DOI:10.1038/nature11344 |
| [6] | Hinohara K, Kobayashi S, Kanauchi H, et al. ErbB receptor tyrosine kinase/NF-κB signaling controls mammosphere formation in human breast cancer. Proc Natl Acad Sci USA, 2012, 109 (17): 6584–6589. DOI:10.1073/pnas.1113271109 |
| [7] | Liao WT, Ye YP, Deng YJ, et al. Metastatic cancer stem cells :from the concept to therapeutics. Am J Stem Cells, 2014, 3 (2): 46–62. |
| [8] | Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis, 2010, 31 (1): 27–36. DOI:10.1093/carcin/bgp220 |
| [9] | Wen B, Wu H, Shinkai Y, et al. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet, 2009, 41 (2): 246–250. DOI:10.1038/ng.297 |
| [10] | Zhang J, Gao Q, Li P, et al. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res, 2011, 21 (12): 1723–1739. DOI:10.1038/cr.2011.176 |
| [11] | Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics :establishment, regulation and biological impact. Mol cell, 2012, 48 (4): 491–507. DOI:10.1016/j.molcel.2012.11.006 |
| [12] | Mosammaparast N, Shi Y. Reversal of histone methylation-biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem, 2010, 79 (7): 155–179. |
| [13] | Tao H, Li H, Su Y, et al. Histone methyltransferase G9a and H3K9 dimethylation inhibit the self-renewal of glioma cancer stem cells. Mol Cell Biochem, 2014, 394 (1-2): 23–30. DOI:10.1007/s11010-014-2077-4 |
| [14] | Sánchez Alvarado A, Yamanaka S. Rethinking differentiation : stem cells, regeneration and plasticity. Cell, 2014, 157 (1): 110–119. DOI:10.1016/j.cell.2014.02.041 |
| [15] | Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell, 2014, 14 (3): 275–291. DOI:10.1016/j.stem.2014.02.006 |
| [16] | Park TS, Donnenberg VS, Donnenberg AD, et al. Dynamic interactions between cancer stem cells and their stromal partners. Current Pathobiology Reports, 2014, 1 (2): 41–52. |
| [17] | Islam F, Qiao B, Smith RA, et al. Cancer stem cell :fundamental experimental pathological concepts and updates. Exp Mol Pathol, 2015, 98 (2): 184–191. DOI:10.1016/j.yexmp.2015.02.002 |
| [18] | Hanahan D, Weinberg RA. Hallmarks of cancer :the next generation. Cell, 2011, 144 (5): 646–674. DOI:10.1016/j.cell.2011.02.013 |
| [19] | Dawson MA, Kouzarides T. Cancer epigenetics :from mechanism to therapy. Cell, 2012, 150 (1): 12–27. DOI:10.1016/j.cell.2012.06.013 |
| [20] | Simon C, Chagraoui J, Krosl J, et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev, 2012, 26 (7): 651–656. DOI:10.1101/gad.186411.111 |
| [21] | Hu S, Yu LL, Li ZM, et al. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biology & Therapy, 2010, 10 (8): 788–795. |
| [22] | Mallen-St Clair J, Soydaner-Azeloglu R, Lee KE, et al. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev, 2012, 26 (5): 439–444. DOI:10.1101/gad.181800.111 |
| [23] | Au SLK, Wong CCL, Lee JMF, et al. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology, 2012, 56 (2): 622–631. DOI:10.1002/hep.25679 |
| [24] | Cai MY, Hou JH, Rao HL, et al. High expression of H3K27me3 in human hepatocellular carcinomas correlates closely with vascular invasion and predicts worse prognosis in patients. Mol Med, 2011, 17 (1-2): 12–20. |
| [25] | Fussbroich B, Wagener N, Macher-Goeppinger S, et al. EZH2 depletion blocks the proliferation of colon cancer cells. PLoS One, 2011, 6 (7): e21651. DOI:10.1371/journal.pone.0021651 |
| [26] | Qi W, Chan H, Teng L, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA, 2012, 109 (52): 21360–21365. DOI:10.1073/pnas.1210371110 |
| [27] | Chase A, Cross NCP. Aberrations of EZH2 in cancer. Clin Cancer Res, 2011, 17 (9): 2613–2618. DOI:10.1158/1078-0432.CCR-10-2156 |