




近年来,实时荧光定量 PCR 技术已被广泛应用于不同生物基因表达的绝对定量和相对定量研究,这项技术因具有高灵敏性、高效性、准确性和重复性好等特点,已成为分子生物学研究领域基因表达分析的重要方法之一,被大量用于分析基因在不同发育时期以及经过不同处理的样本间的表达差异等[1]。
有效的实时荧光定量PCR分析取决于很多因素,如提取的RNA的质量、反转录的效率、引物特异性以及数据处理方法等[2],而采用稳定表达的内参基因来控制实验误差也是非常重要的。理想的内参基因应该是在各种类型的组织、细胞和不同的实验因素条件下均能恒定表达。在真核生物中已经鉴定出一些表达相对稳定的内参基因用于定量表达分析,如广泛使用的β-actin和GAPDH[3],但在原核生物中还没有找到广泛稳定表达的内参基因。大量的研究结果表明,任何一种管家基因的所谓恒定表达都只是在一定类型的细胞或实验因素作用下“有范围”的恒定,并没有表达绝对稳定的基因[4]。Sun等[5]研究了18S rRNA、GAPDH、β-actin和α-tubulin 4个内参基因在茶树中的表达稳定性,发现在不同成熟度的叶片和愈伤组织中表达最稳定的是GAPDH基因,在不同组织器官中表达最稳定的基因则是β-actin。在细菌的基因差异表达分析中,23S rRNA和16S rRNA[6, 7]是经常使用的定量PCR的内参基因,但有研究表明核糖体RNA的表达水平高度依赖于细菌细胞的生理状态,当细胞处于热激、营养匮乏或生长速率发生改变的条件下,核糖体RNA的表达水平发生改变[8, 9],有研究表明,大肠杆菌在碳源[10-12]或无机离子[10, 11]饥饿情况下,16S rRNA发生降解。因此,研究不同的实验材料和不同的实验条件下目标基因的表达时,有必要对内参基因进行重新筛选,并且随着对定量要求的不断提高,为了得到更可靠的实验结果,实验中可选择1个或多个内参基因进行校正[12, 13]。
短小芽孢杆菌(Bacillus pumilus)是一种广泛存在的土壤细菌,在许多生物技术领域有重要的用途: 如工业化生产胞外碱性蛋白酶、拮抗植物病源菌等。开展短小芽孢杆菌的基因表达研究,不仅可以丰富芽孢杆菌的基因表达调控理论,也可为菌株的开发和利用提供重要的指导。要精确分析短小芽孢杆菌的基因表达情况,确定合适的内参基因是前提,但国内外还未见关于其内参基因筛选的报道。为了确定短小芽孢杆菌荧光定量PCR分析的内参基因,我们选择了细菌定量PCR分析中常用的内参基因16S rRNA和rpoB[14-16],以及我们从短小芽孢杆菌SCU11转录组测序数据库中筛选出的3个稳定表达基因(mecA、cadR和sphP),针对短小芽孢杆菌发酵产生碱性蛋白酶的培养过程,采用 geNorm[13]和NormFinder[17]软件对这5个基因的表达情况进行稳定性分析,以期选择出最适于短小芽孢杆菌基因表达水平研究的内参基因。
1 材料与方法 1.1 材料短小芽孢杆菌(Bacillus pumilus)SCU11为本实验室分离并诱变得到的一株高产碱性蛋白酶的菌株。
1.2 方法 1.2.1 菌种活化与发酵培养将保存的菌种接种于LB液体培养基中,37℃振荡培养过夜。活化的菌液以4%的接种量转种于发酵培养基(麸皮2.5%、黄豆粉2%、酵母膏0.3%、K2HPO4 0.4%、NaH2PO4 0.04%及CaCO3 0.3%),转速200 r/min,温度34℃,在发酵培养的不同时间取样。
1.2.2 RNA提取及cDNA合成在菌株生长的不同时期,对应于发酵过程的发酵前期(0 h)、迟缓期(2 h)、对数期(6 h)、转换期(12 h和14 h)、稳定期(24 h)、衰退期(36 h)、酶活最高点(48 h)、酶活下降点(60 h)取样,离心收集菌体,液氮冷冻后于-80℃保存。冻存的菌体取出后加入15 mg/mL的溶菌酶在37℃下温浴10 min,利用TRIzol® Reagent(Life Technologies)提取样品总RNA。RNA的浓度采用Thermos 公司的Nanodrop-2000测定,RNA的完整性通过1%的琼脂糖凝胶电泳检测。
利用PrimeScriptTM RT reagent kit with gDNA Era-ser反转录试剂盒(TaKaRa Biotechnology,Japan)进行反转录,得到的cDNA于-20℃保存,用于荧光定量PCR分析。为了验证RNA样品中是否存在基因组DNA的污染,从反转录之前的反应体系中取出1 μL作为模板进行PCR验证,没有扩增出条带,说明基因组DNA去除完全。
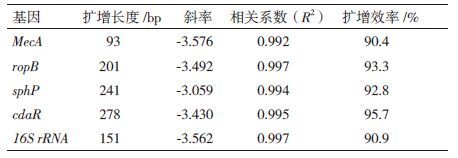
1.2.3 候选内参基因的选择与引物设本研究选择了5个基因进行比较分析: 16S rRNA、mecA、rpoB、cadR和sphP。其中16S rRNA和rpoB是常用的细菌看家基因,mecA、cadR、sphP是我们根据短小芽孢杆菌SCU11转录组数据(结果待发表)中基因的RPKM值筛选而来: 利用RPKM值计算变异系数CV(标准差与平均值的比值)和最大折叠倍数MFC(最大的RPKM值与最小的RPKM值的比值),选择CV值小于4%且MFC值小于2的基因[18]。依据NCBI中短小芽孢杆菌BA06(SCU11的原始野生菌株)基因组中相应基因序列,使用 Primer 5软件设计引物,由成都擎科梓熙生物科技有限公司合成。引物序列,见表 1。

采用 TaKaRa 公司的 SYBR Premix Ex TaqⅡ荧光定量 PCR 试剂盒和 Bio-Rad CFX96 荧光定量PCR仪进行,PCR 反应体系为: 2×SYBR Premix Ex Taq Ⅱ10 µL;cDNA template 2 µL;引物(0.4 µmol/L)各0.5 µL;水7 µL。PCR扩增条件为: 95℃ 1 min;95℃ 5 s,52.5℃ 30 s,72℃ 30 s(在延伸阶段检测荧光强度,收集信号),40个循环;72℃ 10 s。熔解曲线: 加热样品从65-95℃,每隔 0.5℃停留1 s检测1次荧光强度变化。
引物扩增效率验证和标准曲线的绘制: 引物扩增效率(E)与标准曲线的斜率K相关,计算方程式为: E=(10-(1/K)-1)× 100%;制作标准曲线时,将第1链cDNA模板依次稀释5个梯度(1/5、1/25、1/125、1/625和1/3125),研究5个内参基因在发酵的9个不同时间点的表达丰度,每个反应重复3次。解链温度等系列参数通过Bio-Rad CFX 荧光定量PCR 仪自动获得。
1.2.5 数据处理和分析以不同发酵时间点的cDNA稀释液作为实时荧光定量PCR的反应模板,反应体系和条件与标准曲线反应体系相同。将获得的Ct值输入geNorm程序运行,该程序能够计算出每个候选内参基因表达稳定度的平均值(M)并且根据M值的大小进行排序,M值越小,表达就越稳定,从而选出最优内参基因,再通过内参基因的标准化因子的配对差异分析(Vn/n+1)判定内参基因的最适数目。同时利用 NormFinder 程序选出最稳定的基因,二者结合确定最合适的内参基因用于后续实验对目的基因的分析。
1.2.6 目的基因的相对表达分析以mecA和16S rRNA分别作为内参基因,以6 h基因的表达量为校准样本(calibrator),12、24、36、48和60 h的表达量为实验样本(sample),采用2-ΔΔCt相对定量法分析spo0A、sinR和RNA_F_101三个基因的相对表达量。方法如下:
实验样本的△Ct值: △Ct1=Ctsample-Ctref;校准样本的△Ct 值: △Ct2= Ctcalibrator-Ctref;其次,用校准样本的△Ct 值归一试验样本的△Ct 值: △△Ct=△Ct1-△Ct2;最后,计算表达水平比率: 表达量的比值=2-ΔΔCt。
2 结果 2.1 菌体培养及RNA提取在短小芽孢杆菌蛋白酶发酵培养的不同生长时期(0、2、6、12、14、24、36、48和60 h),分别收集菌体采用Trizol法提取RNA。提取的RNA经Nanodrop-2000紫外分光光度计检测浓度和纯度,所有样品的 OD260/OD280的比值均在2.0左右,1%琼脂糖凝胶电泳显示,23S和16S条带亮度清晰(图 1),说明RNA完整性较好。提取的RNA样品再进行去基因组DNA处理,PCR检测表明样品中基因组DNA去除完全。将去除基因组DNA的RNA用于后续的反转录及实时荧光定量 PCR。

|
| 图 1 短小芽孢杆菌SCU11总RNA电泳图谱 |
以梯度稀释的cDNA样本作为模板进行PCR扩增,绘制5个候选内参基因(mecA、rpoB、cadR、sphP、16S rRNA)的标准曲线,得到引物的相关参数(表 2)。表 2显示,5个候选内参基因的线性相关系数(R2)变化范围为0.992-0.997,显示出较好的线性关系。同时根据E =(10-1/slope -1)× 100计算扩增效率,这5个候选内参基因均表现出良好的扩增效率(均大于90%),说明定量结果准确、可靠,符合实时荧光定量PCR对扩增效率的要求。从5个候选内参基因的熔解曲线看(图 2),都只出现单一的信号峰,说明没有引物二聚体和非特异性条带产生。为了进一步证实5个候选内参基因的扩增特异性,将扩增片段经2%的琼脂糖凝胶电泳进行检测,结果(图 3)显示每对引物均扩增出预期大小的片段,且没有出现引物二聚体和非特异性扩增条带,说明引物的特异性较好。

|
| 图 2 候选内参基因的熔解曲线 |

|
| 图 3 候选内参基因扩增产物的琼脂糖凝胶电泳 M: DL1000 Marker;1: mecA;2: rpoB;3: sphP;4: cadR;5: 16S rRNA |
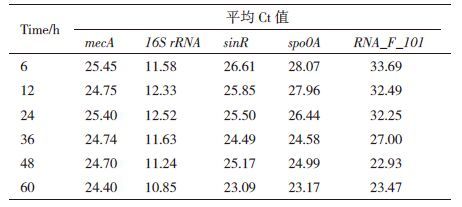
根据5个候选内参基因在不同时间点的平均Ct值(图 4)可以看出,在发酵培养的不同时期5个候选内参基因的表达水平都有一定的变化;其中16S rRNA 作为传统内参基因的表达丰度相对较高,而sphP表达丰度最低。

|
| 图 4 五个候选内参基因在发酵不同时期的表达丰度Ct值 |
通过geNorm软件评估5个候选内参基因在短小芽孢杆菌SCU11不同发酵时期的表达稳定性(M),该软件默认的稳定性值M=1.5,基因的表达稳定值M越小,表明稳定性越高。根据geGorm软件计算,短小芽孢杆菌中5个内参基因在发酵不同生长时期的表达稳定值M排列顺序为sphP(0.099)> rpoB(0.081)> cdaR(0.072)>mecA= 16S rRNA(0.060)(图 5),说明表达最稳定的基因是16S rRNA和mecA。

|
| 图 5 geNorm软件分析各内参基因的表达稳定值M |
在基因的表达分析中,有时需要使用2个或2个以上的内参基因对目的基因进行校正以减少实验误差,从而得到更加准确的结果。利用geNorm程序分析了内参基因的配对差异值 Vn/n + 1,结果(图 6)显示V2/3值为0.024,小于程序推荐值0.15,即无需加入第3个基因进行校正,最合适的内参基因数目是2个,分别是表达最为稳定的mecA和16S rRNA。

|
| 图 6 geNorm软件分析确定用作校准的最适内参基因的数目 |
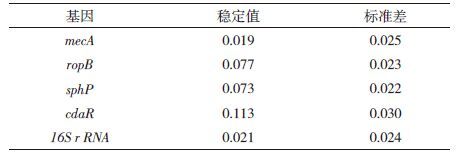
进一步使用NormFinder软件对5个内参基因的表达稳定性进行了分析,M值越小基因表达就越稳定。通过 NormFinder 程序分析荧光定量数据得出每个基因的表达稳定值(M),同时还提供当使用此基因校正时所引用的系统误差值(表 3)。M值的大小顺序为cadR(0.113)>rpoB(0.077)>sphP(0.073)>16SrRNA(0.021)>mecA(0.019)。结果表明,mec-A的M值最小,是表达最稳定的基因。
根据上述的实验结果,在短小芽孢杆菌发酵培养过程中,geNorm软件分析得出16S rRNA和mecA的稳定性最高。NormFinder软件分析的稳定性最高的内参基因是mecA。为了对这两个基因作为荧光定量PCR分析内参基因的可靠性和适应性进行验证,选择sinR、spo0A和RNA_F_101三个基因进行差异表达分析。
结果(表 4)显示,针对我们的cDNA样本,候选内参基因16S rRNA的Ct值在10-13之间,而mecA基因的Ct值在24-26,表明16S rRNA基因的表达水平大大高于mecA基因。从3个目的基因的表达水平以及我们的转录组数据分析来看,绝大多数基因的表达水平更接近于mecA。
分别以mecA和16S rRNA为内参基因,6 h为对照样本,采用2-ΔΔCt相对定量法得出目的基因的相对表达量(表 5)。从表中的数据可以看出,作为一个转录调控因子,sinR在整个发酵过程的表达水平相对比较稳定。另外两个基因RNA_F_101和spo0A,分别用两个内参基因计算出的目的基因表达水平,虽然具体的变化倍数略有变化,但趋势完全一致,说明mecA和16S rRNA基因均可以作为短小芽孢杆菌荧光定量PCR分析的内参基因。

近年来,随着转录组测序、基因表达谱芯片等的迅速发展,实时荧光定量PCR已经成为验证基因表达水平变化的重要技术手段,而选择合适的内参基因做校正是很关键的。理想的内参基因一般应该满足: 选择的内参基因不能为假基因[19];在不同的发育阶段及胁迫条件下表达稳定;表达水平与目标基因相近。然而,不同物种不同生理条件下使用的内参基因各不相同,即使广泛使用的内参基因Actin 和 GAPDH在不同的条件下其表达也会发生改变或受到其他基因的调控[20-23]。因此,研究者需要根据不同的实验条件筛选不同的内参基因。
为了减少选择内参基因的误差,本研究采用了两种不同却又互补的软件进行分析: geNorm和NormFinder,这两种软件被广泛的应用于荧光定量 PCR 中候选内参基因的稳定性评价[24, 25]。geNorm软件是根据候选基因中表达量最高的内参基因与其他候选基因的配对表达水平比值经对数变换,计算其标准差作为基因表达稳定值M,其可以对多个管家基因同时进行比较筛选,选出两个及以上的内参基因,有助于减小系统误差,得到更准确的结果,尤其是对微小表达差异的基因的表达研究具有重要的意义。当然,目的基因有较大表达差异时则不必选择较多的内参基因,这需要视具体实验要求及实验条件而定。NormFinder软件也是常用的一种评估基因表达稳定性的方法,原始数据经△Ct法将其转换成线性表达量,然后通过方差分析得出基因的表达稳定值M,它可以平衡两个变异的来源,但它不能忽略样品制备过程中的系统误差,然而统计方法可以通过大量数据处理来克服这一缺点,所以当不能明确地细分样品时,可以选择这种方法。
目前,国内外还没有对短小芽孢杆菌内参基因筛选及评价的文献报道。在研究短小芽孢杆菌基因表达分析时,应首先考虑选择与目的基因表达水平较为接近的内参基因作为参比。实验利用geNorm和NormFinder两个软件筛选出两个比较稳定的候选内参基因: mecA和16S rRNA。同时,利用这两个候选内参基因对sinR、spo0A和RNA_F_101的表达量进行了分析。sinR基因是一种DNA结合蛋白,它不仅是蛋白酶基因的负调控因子,还参与芽孢生成、鞭毛基因、自溶素生产和感受态的形成等多种细胞内部调控作用[26]。Spo0A是磷酸激活应答调控蛋白家族的成员之一[27],在孢子的初始形成阶段起主要的调控功能。RNA_F_101是我们从短小芽胞杆菌转录组数据库(待发表)中预测出的功能未知的差异表达非编码RNA。结果表明这两个内参基因计算出的3个目的基因的表达水平的变化趋势是完全一致的,说明16S rRNA和mecA都可以作为短小芽孢杆菌荧光定量PCR分析的内参基因。但是,需要注意的是,虽然16S rRNA有较高的表达稳定性,但是由于16S rRNA具有很高的表达水平,其表达量通常显著高于细胞内绝大多数结构基因。在荧光定量PCR实验中,基线通常是3-15个循环的荧光信号,是由于测量的偶然误差引起的,因此,对荧光定量的结果一般取Ct值在15-35个循环的数据进行分析,太大或太小都会导致定量的不准确。在表 4的实验中,16S rRNA的Ct值在11-12左右,如果降低模板浓度将16S rRNA的Ct值控制在理想的15-35个循环之间,目的基因RNA_F_101的Ct值就会大于35个循环,导致结果难以分析。从我们所做的20多个结构基因的荧光定量PCR分析结果以及转录组数据结果来看,大多数基因的表达量都显著低于16S rRNA基因,因此,需要使用一个表达丰度较低的内参基因,mecA就是一个合适的基因。MecA属于MecA 家族蛋白的成员之一,是ClpC六聚体形成及其进一步激活的重要的接头蛋白。当MecA过表达时,会抑制孢子的形成,是孢子形成的负调控因子[28-30]。
4 结论本研究通过geNorm和Normfinder软件对5个荧光定量PCR分析的候选内参基因在短小芽孢杆菌的表达稳定性进行了评估,筛选出稳定的内参基因并通过3个功能基因的差异表达分析进行了验证。结果表明16S rRNA和mecA都可以作为研究短小芽孢杆菌基因表达分析的内参基因,mecA基因由于表达丰度适中,更适合于作为内参基因研究结构基因的表达。为了得到更准确的差异表达结果,也可用两个基因同时进行校正。
| [1] | Huggett J, Dheda K, Bustin S, et al. Real-time RT-PCR normalisation;strategies and considerations. Genes and Immunity , 2005, 6 (4) : 279–284. DOI:10.1038/sj.gene.6364190 |
| [2] | Ginzinger DG. Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Experimental Hematology , 2002, 30 (6) : 503–512. DOI:10.1016/S0301-472X(02)00806-8 |
| [3] | Suzuki T, Higgins P, Crawford D. Control selection for RNA quantitation. Biotechniques , 2000, 29 (2) : 332–337. |
| [4] | 张艳君, 朱志峰, 陆融, 等. 基因表达转录分析中内参基因的选择. 生物化学与生物物理进展 , 2007, 34 (5) : 546–550. |
| [5] | Sun M, Wang Y, Yang D, et al. Reference genes for real-time fluorescence quantitative PCR in Camellia sinensis. Chin Bull Bot , 2010, 45 (5) : 579–587. |
| [6] | Neretin LN, Schippers A, Pernthaler A, et al. Quantification of dissimilatory(bi)sulphite reductase gene expression in Desulfobacterium autotrophicum using real-time RT-PCR. Environmental Microbiology , 2003, 5 (8) : 660–671. DOI:10.1046/j.1462-2920.2003.00452.x |
| [7] | Edwards K, Saunders N. Real-time PCR used to measure stress-induced changes in the expression of the genes of the alginate pathway of Pseudomonas aeruginosa. Journal of Applied Microbiology , 2001, 91 (1) : 29–37. DOI:10.1046/j.1365-2672.2001.01339.x |
| [8] | Vandecasteele S, Peetermans W, Merckx R, et al. Quantification of expression of Staphylococcus epidermidis housekeeping genes with Taqman quantitative PCR during in vitro growth and under different conditions. J Bacteriol , 2001, 183 (24) : 7094–7101. DOI:10.1128/JB.183.24.7094-7101.2001 |
| [9] | Hansen MC, Nielsen AK, Molin S, et al. Changes in rRNA levels during stress invalidates results from mRNA blotting: fluorescence in situ rRNA hybridization permits renormalization for estimation of cellular mRNA levels. J Bacteriol , 2001, 16 : 4747–4751. |
| [10] | Davis B, Luger S, Tai P. Role of ribosome degradation in the death of starved Escherichia coli cells. Journal of Bacteriology , 1986, 166 (2) : 439–445. DOI:10.1128/jb.166.2.439-445.1986 |
| [11] | St John A, Goldberg A. Effects of starvation for potassium and other inorganic ions on protein degradation and ribonucleic acid synthesis in Escherichia coli. Journal of Bacteriology , 1980, 143 (3) : 1223–1233. |
| [12] | Radoni? A, Thulke S, Mackay IM, et al. Guideline to reference gene selection for quantitative real-time PCR. Biochemi Biophys Res Commun , 2004, 313 (4) : 856–862. DOI:10.1016/j.bbrc.2003.11.177 |
| [13] | Vandesompele J, De Preter K, Pattyn F, et al. Accurate normaliza-tion of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology , 2002, 7 : 1–12. |
| [14] | Savard P, Roy D. Determination of differentially expressed genes involved in arabinoxylan degradation by Bifidobacterium longum NCC2705 using real-time RT-PCR. Probiotics and Antimicrobial Proteins , 2009, 1 (2) : 121–129. DOI:10.1007/s12602-009-9015-x |
| [15] | Marco ML, Bongers RS, De Vos WM, et al. Spatial and temporal expression of Lactobacillus plantarum genes in the gastrointestinal tracts of mice. Appl Environ Microbiol , 2007, 1 : 124–132. |
| [16] | Spinsanti G, Panti C, Lazzeri E, et al. Selection of reference genes for quantitative RT-PCR studies in striped dolphin(Stenella coeruleoalba)skin biopsies. BMC Mol Biol , 2006, 7 (1) : 1. DOI:10.1186/1471-2199-7-1 |
| [17] | Andersen CL, Jensen JL, ?rntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Research , 2004, 64 (15) : 5245–5250. DOI:10.1158/0008-5472.CAN-04-0496 |
| [18] | Carvalho DM, de Sá PH, Castro TL, et al. Reference genes for RT-qPCR studies in Corynebacterium pseudotuberculosis identified through analysis of RNA-seq data. Antonie Van Leeuwenhoek , 2014, 106 (4) : 605–614. DOI:10.1007/s10482-014-0231-3 |
| [19] | Nicot N, Hausman JF, Hoffmann L, et al. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. Journal of Experimental Botany , 2005, 56 (421) : 2907–2914. DOI:10.1093/jxb/eri285 |
| [20] | Long XY, Wang JR, Ouellet T, et al. Genome-wide identification and evaluation of novel internal control genes for Q-PCR based transcript normalization in wheat. Plant Molecular Biology , 2010, 74 (3) : 307–311. DOI:10.1007/s11103-010-9666-8 |
| [21] | Mascia T, Santovito E, Gallitelli D, et al. Evaluation of reference genes for quantitative reverse-transcription polymerase chain reaction normalization in infected tomato plants. Molecular Plant Pathology , 2010, 11 (6) : 805–816. |
| [22] | Kou SJ, Wu XM, Liu Z, et al. Selection and validation of suitable reference genes for miRNA expression normalization by quantitative RT-PCR in citrus somatic embryogenic and adult tissues. Plant Cell Reports , 2012, 31 (12) : 2151–2163. DOI:10.1007/s00299-012-1325-x |
| [23] | 周兰, 张利义, 张彩霞, 等. 苹果实时荧光定量PCR分析中内参基因的筛选. 果树学报 , 2012, 29 (6) : 965–970. |
| [24] | 黄雪玲, 冯浩, 康振生. 小麦条锈菌实时荧光定量PCR分析中内参基因的选择. 农业生物技术学报 , 2012, 20 (2) : 181–187. |
| [25] | 薛承美, 解廷娜, 叶素丹, 等. 利用实时荧光定量PCR筛选新蚜虫疠霉内参基因. 农业生物技术学报 , 2014, 22 (12) : 1575–1583. |
| [26] | Bai U, Mandic-Mulec I, Smith I. SinI modulates the activity of SinR, a developmental switch protein of Bacillus subtilis, by protein-protein interaction. Genes & Development , 1993, 7 (1) : 139–148. |
| [27] | Ferrari FA, Trach K, LeCoq D, et al. Characterization of the spo0A locus and its deduced product. Proceedings of the National Academy of Sciences , 1985, 82 (9) : 2647–2651. DOI:10.1073/pnas.82.9.2647 |
| [28] | Boutry C, Wahl A, Delplace B, et al. Adaptor protein MecA is a negative regulator of the expression of late competence genes in Streptococcus thermophilus. Journal of Bacteriology , 2012, 194 (7) : 1777–1788. DOI:10.1128/JB.06800-11 |
| [29] | Wang F, Mei Z, Qi Y, et al. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature , 2011, 471 (7338) : 331–335. DOI:10.1038/nature09780 |
| [30] | Tian XL, Dong G, Liu T, et al. MecA protein acts as a negative regulator of genetic competence in Streptococcus mutans. Journal of Bacteriology , 2013, 195 (22) : 5196–5206. DOI:10.1128/JB.00821-13 |