



2. 军事医学科学院微生物流行病研究所, 北京 100071
2. Institute of Microbiology and Epidemiology, the Academy of Military Medical Sciences, Beijing 100071
高通量测序技术的发展,揭示了人类皮肤表面(尤其是手掌面皮肤上)微生物(细菌)群落的组成有丰富的多样性[1-7]。不同种族之间,由于受生活方式、环境条件等因素的影响,皮肤微生物群落的结构组成差异明显[8-10]。不同家庭的人之间其皮肤微生物群落的差异大于同一家庭内部,而同一家庭内部,不同个体之间的皮肤微生物群落特征存在差异性[11]。人手掌表面微生物的群落组成受性别、左右手习惯、洗手后时长的影响[12]。不同的个体其皮肤微生物群落结构随时间变化会产生一定的变动,而其变动规律、变化范围也是因人而异的,在个体间存在明显差异性[13]。
高通量测序技术实验环节步骤较多,每一步骤细微的差别都可能在最终的测序结果中放大,并造成测序结果、分析结果与实际情况的偏差[14]。首先,在提取微生物群落DNA的过程中,由于不同种类的细菌其细胞壁结构不同,而不同的DNA提取试剂盒裂解细胞、吸附DNA的原理、方法又各不相同,造成提取效率、提取质量上的差异。其次,在PCR扩增过程中,又存在多种引起PCR产物成分变化的因素,如不同引物对不同DNA模板的偏好性不同、退火温度改变时也会引起扩增效率的变化等等。再次,在测序过程中,使用不同的测序平台、样品的测序深度不同时,也会出现分析结果与实际情况的差异。
最新的研究显示,实验设计会显著地影响皮肤微生物组的分析结果。宏基因组shotgun测序与基于16S rRNA基因测序相比各有优缺点。宏基因组shotgun测序可用于分析微生物群落的代谢途径和基因功能,但其应用受限于微生物群落DNA含量和参考基因组。一方面,在微生物群落DNA含量较低时,往往无法达到测序要求;另一方面,由于环境微生物中大多数微生物的序列仍未知,所以宏基因组数据中高达90%的序列由于缺少参考序列而不能被确定[15]。16S rRNA基因的测序结果由于有丰富参考序列可供比对,因此在物种识别上有很大优势。但由于16S rRNA基因并非单拷贝基因,即使在同一属的不同种之间拷贝数都可能存在差异,因此其测序结果总体上来说仅仅是半定量的[16],会使微生物群落的α多样性及β多样性的分析结果出现偏差。16S rRNA基因的通用引物的不同,会导致微生物群落结构特征分析结果的不同。这是因为用于通用引物设计的保守区间,在亲缘关系较远的物种之间可能保守性不足,导致某些种属的细菌扩增效率差,从而使得微生物群落的α多样性及β多样性的分析结果出现偏差。与宏基因组shotgun测序相比,实验16S rRNA基因的V1-V3区测序获得的微生物群落结构分析结果与之相似,而V4区测序获得的微生物群落结构分析结果却不尽人意[17]。在高通量测序过程中,模板的含量高低会对测序结果造成显著影响[18],因此研究各个实验因素对此类实验最终结果的影响是非常必要的。
由于皮肤表面的微生物的含量与土壤、粪便等环境微生物相比含量较低,因此使用高通量测序技术分析皮肤表面的微生物群落结构特征时,更容易受到实验过程中各个环节的影响,有必要对样品采集、DNA提取、PCR扩增、测序等各个实验环节的条件进行筛选优化。本研究旨在通过实验设计和数据分析,筛选出尽可能真实反映含量较低的皮肤微生物群落的高通量测序分析方法。
1 材料与方法 1.1 材料 1.1.1 实验菌种来源本论文实验所用细菌种类共计15种,包括蜡样芽孢杆菌(Bacillus cereus)、摩氏摩根菌(Morganella morganii)、脱氮假单胞菌(Pseudomonas denitrificans)、弗氏柠檬酸杆菌(Citrobacter freundii)、鲍曼不动杆菌(Acinetobacter baumannii)等,所有菌种均由军事医学科学院微生物流行病研究所提供。
1.1.2 主要试剂和仪器BactoTM Brain Heart Infusion(Becton,Dickinson and Company Sparks,美国),细菌DNA提取试剂盒(杭州新景生物试剂开发有限公司,中国),PowerSoil DNA Isolation Kit(MO BIO Laborotorise Inc,美国),High Pure PCR Template Preparation Kit(Roche Diagnostics GMbH,德国),H2O3-PRO 金属浴(金银杏生物科技有限公司,中国),JY88-Ⅱ DN 超声波信号发生器(南京新辰生物科技有限公司,中国),TissueLyser Ⅱ(QIAGEN,德国),Gene Amp PCR System 9700(Applied Biosystems,美国),Qubit®2.0 Fluorometer(Life technologies,美国),Ion Torrent Personal Genome Machine(Life Technolo-gies,美国)。
1.2 方法 1.2.1 细菌菌种的培养及计数菌种从-70℃冰箱取出后,于BHI固体培养基上划线,放置于37℃培养箱中培养。菌落形成以后挑取单克隆于BHI液体培养基中,37℃空气摇床中培养至对数期。然后将菌液按梯度稀释后,选取稀释度为10-7、10-8、10-9的菌液100 μL涂布于BHI固体培养基上(每个稀释梯度设置3个重复),放置于37℃培养箱中培养,菌落形成后计数。
1.2.2 影响提取细菌群落DNA效率的因素分析挑选菌液浓度在1.0×108个/mL以上的细菌,稀释至1.0×108个/mL的浓度,然后等比例混合,形成浓度为1.0×108个/mL的模拟细菌群落。并选用一定浓度1.0×108个/mL的模拟细菌群落,将浓度稀释为1.0×107个/mL的模拟细菌群落。
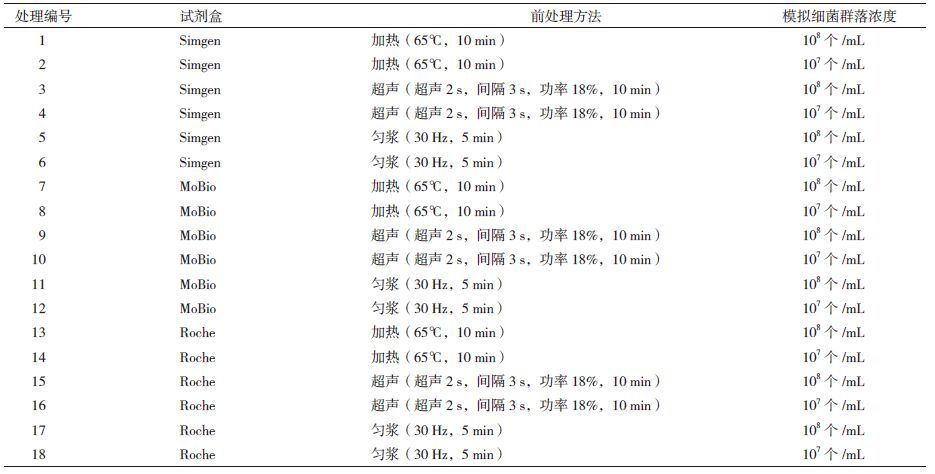
实验设计为有重复的3因素混合水平实验,主要考查3种DNA提取的前处理方法(65℃加热处理10 min、超声2 s,间隔3 s,功率18%,处理10 min、30 Hz匀浆处理5 min)、3种DNA提取试剂盒(SimGen、MoBio、Roche)在提取两种浓度不同(1.0×108个/mL、1.0×107个/mL)的模拟细菌群落时获得的DNA质量、数量(表 1)。本部分实验做了3次重复,分别标记为A组、B组、C组,提取到的DNA使用1%琼脂糖凝胶,140 V电压下电泳30 min后凝胶摄像。A组、B组、C组提取到的DNA均使用Qubit 2.0荧光定量。
为了尽可能减小引物偏好性对最终测序结果的影响,本部分实验所用模拟细菌群落模板制备方法为分别使用单克隆菌液提取DNA,Qubit 2.0定量后统一将稀释为2 ng/μL,等比例混合之后梯度稀释为1、0.5、0.25和0.125 ng参与PCR反应。
实验设计为无重复的两因素混合水平实验,主要考查模板量(4、2、1、0.5、0.25和0.125 ng)和退火温度(55℃、60℃、65℃)2个因素对测序结果的影响。本部分实验选取引物为皮肤微生物组研究中使用广泛的扩增16S rRNA基因V1-V2区的通用引物:F27(5'-AGAGTTTGATCCTGGCTCAG-3'),R338(5'-TGCTGCCTCCCGTAGGAGT-3')。PCR体系为50 μL体系:Q5 High-Fidelity 2×Master Mix 25 μL,上下游引物(每个处理所用引物均在5'端带有4 bp barcode,引物浓度为10 pmol)各1 μL,模板1 μL,ddH2O 22 μL。PCR条件:预变性95℃ 5 min;变性95℃ 30 s,退火55℃/60℃/65℃ 30 s,延伸72℃ 30 s,25个循环;延伸7 min。该部分实验根据退火温度的不同分为3组(标记为D组、E组、F组),同时在3台9700 PCR仪上扩增,每组设置一个阴性对照,不添加模板DNA。PCR扩增产物经0.1%琼脂糖凝胶电泳检测、Qubit 2.0荧光定量,浓度大于10 ng/μL的PCR产物建库后使用Ion torrent PGM测序仪测序。
1.2.4 影响采集细菌群落DNA效率的因素分析将菌液分别稀释至107个/mL,等比例混匀后充分涡旋震荡,形成107个/mL的模拟群落。吸取100 μL混合模拟群落的菌液,滴于载玻片上,待其完全干燥后,用于该部分采集实验(实验操作均在超净工作台内进行)。
实验设计为三因素混合水平实验,主要考查3种样品采集工具(圆头棉签、尖头棉签、尼龙签)、3种前处理方法(加热、超声、匀浆)、2种采集液体(PBS缓冲液、0.1% Tween 20+0.15 mol/L NaCl)在采集微量模拟细菌群落(106个)时,会对测序结果产生怎样的影响。本部分实验设置为G、H两组,G组实验使用Roche High Pure PCR Template Preparation Kit提取,H组实验使用Mo Bio PowerSoil DNA Isolation Kit提取,然后根据Ⅱ部分实验结果选用最优的PCR条件扩增16S rRNA基因V1-V2区。PCR扩增产物经0.1%琼脂糖凝胶电泳检测、Qubit 2.0荧光定量,浓度大于10 ng/μL的PCR产物建库后使用Ion torrent PGM测序仪测序。
1.2.5 测序深度对皮肤微生物组结构特征分析的影响分析样品采集自一名29岁健康女性左右两手手掌部皮肤,在其洗手3 h后采样(因为洗手会引起皮肤表面细菌群落的改变)。
测序平台选择目前应用广泛的Ion Torrent Personal Genome Machine,测序深度设置为10000条/样品、20 000条/样品及40 000条/样品。样品采集方式、DNA提取方法、PCR反应条件均选用前三部分实验筛选出的条件。PCR扩增产物经0.1%琼脂糖凝胶电泳检测、Qubit 2.0荧光定量后建库、测序。测序结果使用QⅡME1.8.0软件分析。
2 结果 2.1 细菌菌种的培养及计数细菌菌落计数结果及推算的细菌菌液浓度见表 2。
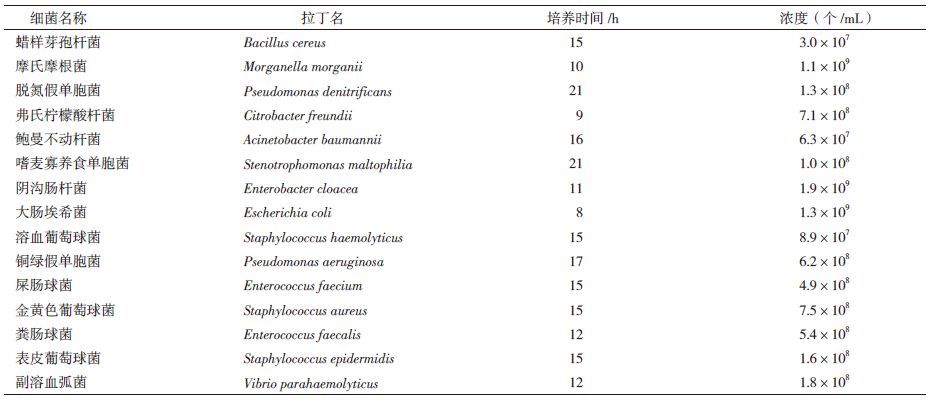
图 1为A组实验重复的电泳图,从图中可以看出,3种试剂盒中,Simgen试剂盒提取的DNA含量最低,电泳检测的条带微弱甚至没有;MoBio试剂盒在配合各种前处理方法时均能得到整齐的基因组DNA条带;而Roche试剂盒在配合各种前处理方法时除了能提取出基因组DNA,同时还能提取到其他一些小片段DNA。B组实验重复和C组实验重复所获电泳图检测结果与A组类似。

|
| 图 1 A组重复DNA提取结果电泳图 |
对A、B、C三组重复实验获得的DNA浓度测量值使用SPSS 21.0进行方差分析和多重比较分析,发现试剂盒种类和模拟细菌群落的浓度对最终DNA提取的浓度有极显著影响,而不同的前处理方法对最终DNA提取的浓度没有显著影响。多重比较分析显示,Roche试剂盒相对于Simgene试剂盒存在极显著差异,而Roche试剂盒与MoBio试剂盒、MoBio试剂盒与Simgene试剂盒之间存在显著差异;模拟细菌群落中不同细菌浓度之间(1.0×108个/mL、1.0×107个/mL)也存在极显著差异。综合考虑3种不同的试剂盒在DNA提取的数量和质量上的结果,决定选取Roche试剂盒和MoBio试剂盒进入下一阶段的实验。考查不同的试剂盒及前处理方法对测序结果的影响。
2.3 影响模拟细菌群落PCR扩增效率的因素分析测序结果使用CLC DNA Workbench处理分析,统计各个种类细菌16S rRNA基因序列的条数。模拟细菌群落中,由于各种细菌DNA的总量、基因组DNA大小、以及16S rRNA基因拷贝数已知,因此各种细菌的16S rRNA基因拷贝总数及比例可以计算得出。使用SPSS 21.0软件对实验统计结果和模拟群落的细菌组成比例相似性进行分析,使用Euclidean 距离(0.142)、Chebychev距离(0.070)、平方 Euclidean 距离(0.020)等方法分析的结果均显示,在退火温度为55℃、模板浓度为2 ng的条件下,PCR产物的测序结果中各种细菌的比例与实际投入的模拟群落的细菌组成比例最为接近。
2.4 影响采集细菌群落DNA效率的因素分析测序结果使用CLC DNA Workbench处理分析,统计各个种类细菌16S rRNA基因序列的条数。由于各种细菌的数量,以及16S rRNA基因拷贝数已知,各种细菌的16S rRNA基因拷贝总数及比例可计算得出。使用SPSS 21.0软件对统计结果和模拟群落的细菌组成比例相似性进行分析,使用Euclidean 距离(0.276)、Chebychev距离(0.179)、平方 Euclidean 距离(0.076)等方法分析的结果均显示,选用尖头棉签或尼龙签,蘸取0.1% Tween 20+0.15 mol/L NaCl采集样品后,经过超声前处理,再使用Roche High Pure PCR Template Preparation Kit提取DNA,能使得PCR产物的测序结果中各种细菌的比例与实际投入的模拟群落的细菌组成比例最为接近。
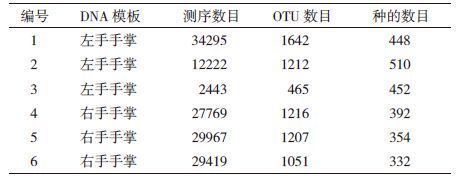
2.5 测序深度对皮肤微生物组结构特征分析的影响分析测序结果使用QⅡME 1.8.0软件进行分析。选择质量>20且长度>200 bp的序列进行分析,根据97%的序列相似性进行OTU的划分。结果(表 3)显示,在序列条数少于10 000条时,随着测序深度增加,样品中可检测到的OTU种类呈迅速增加的趋势,但当测序序列的数目在10 000-20 000条之间时,检测到OTU种类的增长较为缓慢。测序数据达到30 000条之后就趋于平台期。因此,在分析此类样品时,测序序列选择在20 000-30 000条之间较为合适。将所有样品中获得的OTU种类的数目进行统计,占比例最高的4个属分别是Propionibacterium、Corynebacterium、Staphylococcus及Ochrobactrum(图 2,编号同表 2)。

|
| 图 2 不同样品在属一级上的相对丰度 |
使用加权unifrac距离对不同测序深度的样品间的群落组成相似度进行分析。结果显示,不同测序深度的左手手掌的样品间加权unifrac距离为0.086 717-0.154 386,而不同重复的右手手掌的样品间加权unifrac距离为0.084 148-0.142 199,左右两只手掌样品间的加权unifrac距离为0.130 406-0.246 853。因此,不论测序深度存在怎样的差异,左右两只手掌间的皮肤微生物组群落组成还是存在显著差异的。基于加权unifrac距离的UPGMA建树(图 3,编号同表 3)显示自两只手掌的样品各自聚类成一枝。

|
| 图 3 基于加权unifrac距离的UPGMA建树 |
DNA测序技术的发展确立了宏基因组学在科学研究领域中的地位,但由于第一代测序方法存在低通量的原因,限制了所能研究的样品的物种复杂度[19, 20]。利用新一代测序技术对来自大量手掌皮肤样本的研究,获得了更深度的DNA样品序列信息[21, 22]。极大促进了更深入的挖掘人类皮肤微生物的种类,拓展了人们对皮肤微生物群落组成的认识。因此,新一代高通量测序技术促进了皮肤微生物组群落结构应用于法庭科学人身同一认定的发展。Fierer等[12]研究了51名志愿者双手上的皮肤微生物群落,平均每人每只手上有156种系统发育型(phylotype)。在本实验中,随着测序深度的增加,手掌面皮肤上细菌OTU的种类平均可达510种,说明测序深度的增加会使得皮肤生物群落的分析更加详尽。
美国印第安人Yanomami部落是与外界接触非常少的一个族群,其皮肤微生物组的优势属为Staphylococcus、Corynebacterium、Neisseriaceae及Propionibacterium[9]。Leung等[10]的研究结果显示,黄种人皮肤上细菌的优势属主要是Propionibacterium、Corynebacterium、Staphylococcus及Enhydrobacter,且黄种人手掌部皮肤微生物群落的结构组成与美国白种人有显著差异。在本文的实验中,从志愿者手掌皮肤上所取样品里占比例最高的4个属分别是Propionibacterium、Corynebacterium,Staphylococcus及Ochrobactrum,与Leung等的结果基本一致。
4 结论经过实验设计和数据分析,优化筛选出的适合于分析人类皮肤微生物组的实验方法如下:选用尖头棉签或尼龙签,蘸取0.1% Tween 20+0.15 mol/L NaCl采集样品,超声处理后使用Roche High Pure PCR Template Preparation Kit试剂盒提取DNA,PCR退火温度为55℃,模板浓度为2 ng时,16S rRNA基因V1-V2区的测序数据分析的细菌群落组成结果与模拟细菌群落中各种细菌的含量比例最为接近。测序深度达到20 000-30 000条时即可以满足后续细菌群落多样性分析的要求。
| [1] | Petrosino JF, Highlander S, Luna RA, et al. Metagenomic pyrosequencing and microbial identification. Clinical Chemistry , 2009, 55 (5) : 856–866. DOI:10.1373/clinchem.2008.107565 |
| [2] | Grice EA, Segre JA. The skin microbiome. Nature Reviews Microbiology , 2011, 9 (4) : 244–253. DOI:10.1038/nrmicro2537 |
| [3] | Fierera N, Lauberb CL, Zhoub N, et al. Forensic identification using skin bacterial communities. Proc Natl Acad Sci USA , 2010, 107 (14) : 6477–6481. DOI:10.1073/pnas.1000162107 |
| [4] | Caporaso JG, Lauber CL, Costello EK, et al. Moving pictures of the human microbiome. Genome Biology , 2011, 12 : R50. DOI:10.1186/gb-2011-12-5-r50 |
| [5] | Kueneman JG, Parfrey LW, Woodhams DC, et al. The amphibian skin-associated microbiome across species, space and life history stages. Molecular Ecology , 2014, 23 (6) : 1238–1250. |
| [6] | Tomic-Canic M, Perez-Perez GI, Blumenberg M. Cutaneous microbiome studies in the times of affordable sequencing. Journal of Dermatological Science , 2014, 75 (2) : 82–87. DOI:10.1016/j.jdermsci.2014.05.001 |
| [7] | Scharschmidt TC, Fischbach MA. What Lives on our skin:ecology, genomics and therapeutic opportunities of the skin microbiome. Drug Discov Today Dis Mech , 2013, 10 (3-4) : Pii:e83–e89. DOI:10.1016/j.ddmec.2012.12.003 |
| [8] | Blaser MJ, Dominguez-Bello MG, Contreras M, et al. Distinct cutaneous bacterial assemblages in a sampling of South American Amerindians and US residents. International Society for Microbial Ecology , 2013, 7 : 85–95. |
| [9] | Clemente JC, Pehrsson EC, et al. The microbiome of uncontacted Amerindians. Sci Adv , 2015, 1 (3) : pii:e1500183. DOI:10.1126/sciadv.1500183 |
| [10] | Leung MHY, Wilkins DH and Lee P K. Insights into the pan-microbiome:skin microbial communities of Chinese individuals differ from other racial groups. Sci Rep , 2015, 5 : 11845. DOI:10.1038/srep11845 |
| [11] | Song SJ, Lauber C, Costello EK, et al. Cohabiting family members share microbiota with one another and with their dogs. eLife , 2013, 2 : e00458. |
| [12] | Fierera N, Hamadyc M, et al. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA , 2008, 105 (46) : 17994–17999. DOI:10.1073/pnas.0807920105 |
| [13] | Flores GE, Caporaso JG, Henley JB, et al. Temporal variability is a personalized feature of the human microbiome. Genome Biology , 2014, 15 : 531. DOI:10.1186/s13059-014-0531-y |
| [14] | Meisel JS, Hannigan GD, Tyldsley AS, et al. Skin microbiome surveys are strongly influenced by experimental design. J Invest Dermatol , 2016, 136 (5) : 2016. |
| [15] | Huson DH, Richter DC, et al. Methods for comparative metageno-mics. BMC Bioinformatics , 2009, 10 (Suppl 1) : S12. DOI:10.1186/1471-2105-10-S1-S12 |
| [16] | Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur:open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology , 2009, 75 : 7537–7541. DOI:10.1128/AEM.01541-09 |
| [17] | Youssef N, Sheik CS, Krumholz LR, et al. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Applied and Environmental Microbiology , 2009, 75 : 5227–5236. DOI:10.1128/AEM.00592-09 |
| [18] | Kennedy K, Hall MW, Lynch MDJ, et al. Evaluating bias of illumina-based bacterial 16S rRNA gene profiles. Applied and Environmental Microbiology , 2014, 80 (18) : 5717–5722. DOI:10.1128/AEM.01451-14 |
| [19] | Grice EA, Kong HH, Renaud G, et al. A diversity profile of the human skin microbiota. Genome Res , 2008, 18 : 1043–1050. DOI:10.1101/gr.075549.107 |
| [20] | Ursell LK, Clemente JC, Rideout JR, et al. The interpersonal and intrapersonal diversity of human associated microbiota in key body sites. J Allergy Clini Immunol , 2012, 129 (5) : 1204–1208. DOI:10.1016/j.jaci.2012.03.010 |
| [21] | Liu Z, Lozupone C, Hamady M, et al. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Research , 2007, 35 (18) : e120. DOI:10.1093/nar/gkm541 |
| [22] | Hamady M, Walker JJ, Harris JK, et al. Error-correcting barcoded primers allow hundreds of samples to be pyrosequenced in multiplex. Nature Methods , 2008, 5 (3) : 235–237. DOI:10.1038/nmeth.1184 |