




2.广东第二师范学院生物与食品工程学院,广州 510303
2. School of Biology and Food Engineering,Guangdong University of Education,Guangzhou 510303
双花木属(Disanthus)隶属金缕梅科(Hamam-elidaceae),现存双花木原种(D. cercidifolius)和亚种长柄双花木(D. cercidifolius subsp. Longipes),由于分布区域生境破坏严重,自然居群大小正骤减,被列为国家二级重点保护濒危物种。该属植物为落叶小乔木或灌木,并且是一种优良的(观叶、花和果)珍稀观赏植物[1]。其叶为宽卵圆形,基部心形,后期转红色,长柄双花木因叶柄长而得名;花序头状,腋生,有花5-8朵,排在一个平面上;果是蒴果,种子黑色,室间开裂,每室有光亮的种子数颗。该属植物大多分布于山体阴坡或半阴坡,零星生长于低山丘陵地、山洼谷底滩地、沟谷溪流边或溪边坡上常绿与落叶阔叶混交林林缘,树高约为2-5 m。双花木自然地理分布区仅残存于日本南部本州和四国的山地,长柄双花木为我国中亚热带中东部特有,仅分布于广东与广西北部、湖南、江西及浙江西部的山地,近年在粤、桂北和赣东北发现新居群[2, 3]。对双花木属染色体和核型的分析结果表明[4, 5],双花木的染色体为2n=16,潘开玉与杨亲二[6]的研究结果进一步得出中国的与日本的核型差异十分明显,但是形态学的差异很小,因此两个亚种的分类地位存在争议。Shi等[7]通过基因序列检测来探讨金缕梅科的系统发育(宏观进化)关系,认为双花木属与红花荷属、壳菜果属、马蹄荷属等近缘,而与形态性状不太一致[8, 9, 10, 11]。近年来,随着DNA测序技术的迅猛发展,对植物基因组研究的不断深入,DNA序列以其独特的优势被广泛应用于谱系地理研究。对于植物而言,叶绿体cpDNA和核糖体rDNA常被用于属及属以下分类单元的研究,其中叶绿体DNA的非编码区序列(内含子和基因间隔区)和核糖体DNA内转录间隔区(ITS)[12, 13]因其功能上的限制较少,所以进化速率比编码区快,因此很适合用于种内系统发育关系研究。本研究从分子遗传学的角度研究双花木属的系统发育关系,为后续深入研究双花木属的谱系地理等科学问题提供参考依据。
1 材料与方法 1.1 材料本实验所用样本均来自双花木属在自然分布区采集的16个居群共384个个体的叶片样本,每个居群24个个体,分别采集新鲜嫩叶,分装于装有硅胶塑料袋内进行快速烘干,并带回实验室冰箱保存。

采用改良的CTAB法[14]提取叶片总DNA,PCR引物序列如下:
rps16F:5'-GTGGTAGAAAGCAACGTGCGACTT-3';rps16R2:5'-TCGGGATCGAACATCAATTGCA-AC-3'[15]。
psbA:5'-GTTATGCATGAACGTAATGCTC-3';trnH:5'-CGCGCATGGTGGATTCACCCAATCC-3'[16]。
psaA-ycf3-F:5'-CATTCCTCGAACGAAGTTTTTA-CGGGATCC-3';psaA-ycf3-R:5'-TCCCGGTAATTATA-TTGAAGCGCATAATTG-3'[17]。
PCR反应在PTC-200(MJ Research,USA)热循环仪上来进行。20 μL PCR反应体系如下:10 μL 2×Taq PCRMixC(北京博迈德生物技术有限公司),DNA模板1 μL,引物0.2 μL,加ddH20补至20 μL。反应程序为:94℃预变性5 min;94℃加热变性30 s,55℃退火1 min,72℃延伸1 min,循环35次;最后在72℃延伸10 min。
扩增产物在1.8%琼脂糖凝胶上检测,用溴化乙锭染色,在凝胶成像系统下观察有无扩增产物并拍照留记录,若扩增电泳条带整齐单一,亮度较高,则将扩增产物送到北京华大基因生物有限公司进行纯化并测序。部分样本引物扩增效果,见图 1。

|
| 图 1 长柄双花木部分个体cpDNA psaA-ycf3序列扩增产物电泳图 |
测序获得的序列先用contig软件进行正反拼接,获得原始数据,再用clustalX软件进行序列比对,对有差异的位点在Chromas软件中对应原始峰形图确认是否为真实变异位点,最后在BioEdit 7.0.9[18]软件对不准确位点进行手工校正,保存获得最终数据文件。利用DnaSP5.0[19]软件对cpDNA单倍型、序列变异位点(Polymorphic)以及简约信息位点(Parsimony informativesites)等参数进行统计。用DIVA-GIS7.5.0软件和ArcMaplO绘制单倍型分布图。

|
| A-H1;B-H2;C-H3;D-H4;E-H5;F-H6;G-H7;H-H8;I-H9;J-H10;K-H11 图 2 双花木属样品居群和cpDNA单倍型分布图 |
用软件PERMUT计算居群cpDNA序列的遗传多样性(Ht)和居群内平均遗传多样性(Hs),居群间遗传分化系数Gst以及Nst值,计算时进行1 000次的置换检验。计算Gst值时只考虑单倍型频率,其计算公式为Gst=(Ht-Hs)/Ht。用ARLEQUIN 3.0[20]软件包中的分子变异分析(AMOVA)检测组间、组内居群间和居群内的遗传差异以及居群内,居群间单倍型分布的分子变异分析(Fst)[21],并在ARLEQUIN中计算每个居群的单倍型多态性(Hd)和核苷酸多态性(π)[22]。
1.2.4 系统发育分析用PAUP4.0聚类和Bayesian方法对所得叶绿体单倍型进行系统发育关系分析。先用MODELTEST 3.06[23]搜索最适合当前数据的模型和各种参数。在PAUP*4.0bl0[24]软件下,构建最大简约树(Most parsimony,MP),其中,空位或缺失在分析之前进行替代处理。在MrBayes软件[25]分析中,在一条冷链和连续递增的三条热链下共运行了1×107代,其中每2 500代保存一颗树,最后获得4 000颗树,本文中在计算多数一致性树及各分枝的后验概率均舍弃了前2 500棵树。
2 结果 2.1 cpDNA序列变异和居群分布从双花木属自然地理分布区16个居群的164个个体中检测了3个cpDNA片段,分别为:psbA-trnH、rps16F-R2和psaA-ycf3。序列经比对后长度分别为:321 bp、601 bp及611 bp,其中psbA-trnH片段检测出3种单倍型,rps16F-R2片段检测出6种单倍型,psaA-ycf3片段检测出5种单倍型。基于cpDNA无重组、单亲遗传的特点,我们可以把3个叶绿体片段串联起来分析,串联后共检测出11种单倍型(H1-H11)(表 2),串联序列总长度为1 553 bp,包含22个变异位点,其中12个是简约信息位点。
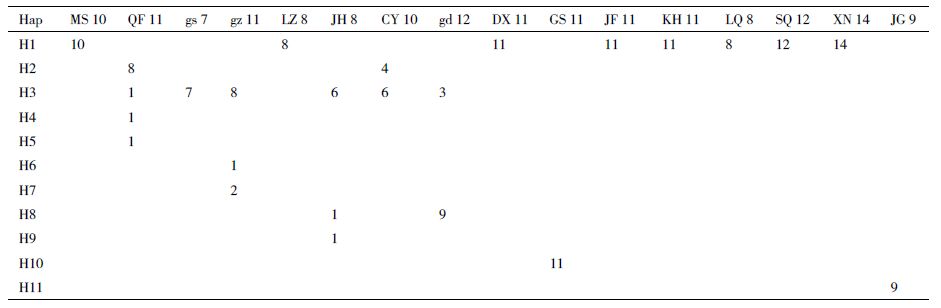
分析得到双花木属所有序列单倍型多态性(Hd)为0.679 77,总核昔酸多态性(π)为0.002 52。检测的11种单倍型中,H1和H3为广布单倍型(图 3),其中单倍型H1分布最广,中国的长柄双花木大多数居群共享这种单倍型,为85个个体所共有。此外,日本的双花木群体除了gs居群只有单一的一种单倍型外,其他居群都检测到两种或两种以上的单倍型;中国的长柄双花木群体则所有居群都只检测到单一的一种单倍型,且除了官山(GS)和井冈山(JG)有独享单倍型外,其他居群都共享单倍型H1。

|
| 图 3 基于PAUP和MrBayes的双花木属cpDNA系统发育树 |
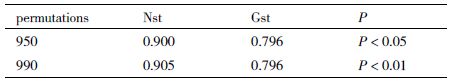
双花木属每个居群的cpDNA单倍型多态性(Hd)、核苷酸多态性(π)及核苷酸差异数(k)等遗传多样性参数见表 3。结合表 2可以看出,日本的居群QF的单倍型种类最多,有4种单倍型(H2、H3、H4和H5),其单倍型多态性(Hd)为0.490 91。在所有居群中cpDNA遗传多样性(Hd)范围在0-0.533 33,核苷酸多态性(π)的范围为0-0.002 01,最高的单倍型多态性和核苷酸多态性存在于日本本州岛中部的CY居群和JH居群中,分别为0.533 33和0.002 01。在所有16个居群中,居群间总的遗传多样性Ht(0.725)高于居群内平均遗传多样性场Hs(0.148)。由表 4可知,居群间遗传分化系数Gst为0.796,在1 000次置换检验中,950次的置换结果Nst(0.900)大于Gst,即Nst>Gst(P<0.05),说明亲缘关系较近的单倍型发生在同样居群中的概率较高,且存在着显著的谱系地理学结构;在1 000次置换检验中,990次的置换结果Nst(0.905)也大于Gst,即Nst>Gst(P<0.01),说明双花木属植物亲缘关系较近的单倍型发生在同样居群中的概率极高,且存在着极显著的谱系地理学结构。基于cpDNA单倍型的AMOVA分析结果(表 5)显示,双花木属的变异主要来自群体间,即中国和日本的两个群体分化明显,变异占53.08%,组内居群间的变异占24.11%,来自个体间的变异为22.80%。


本研究选用蕈树(Altingia chinensis)为外类群,是与双花木属同为金缕梅科的蕈树属(Altingia),基于PAUP的最大简约法(Most parsimony tree,MP)和贝叶斯模型(Bayesian)构建系统发育树,利用1 000次重复的自展分析做可靠性检验,其中,空位或缺失在分析之前进行替代处理。两种方法构树得到的拓扑结构基本一致,结构如图 3所示。从图中可以看到双花木属分为明显的中国和日本两大支系,一支来自中国居群所属的单倍型H1、H10和H11,MP支持率为86%,Bayesian后验概率PP值为1;一支来自日本居群所属的单倍型H2-H9,MP支持率为95%,Bayesian后验概率PP值为1。其中,日本的双花木群体又分为两组支系,支系的MP支持率为80%,Bayesian后验概率PP值为1。两组支系一组来自日本本州岛的中部(居群QF、CY),所在单倍型为H2和H5;另一组来自日本本州岛的西南部(居群gz、gd和JH),所在单倍型为H3、H4和H9。中国的长柄双花木则所有居群拥有的单倍型以平行单支系的方式聚在一起,系统发育结果揭示了双花木属植物中国与日本两亚种间发生了遗传分化。
3 讨论 3.1 居群遗传多样性及遗传结构本研究中通过对双花木属cpDNA 3个基因片段序列的联合分析,得到双花木属总单倍型多态性(Hd)为0.679 77,总核苷酸多态性(π)为0.002 52,日本群体的遗传多样性高于中国群体,单倍型多态性和核苷酸多态性最高的居群存在于日本本州岛的中部的CY居群和JH居群,分别为0.533 33和0.002 01,结合野外采集样品的调查结果,分析造成这一结果的原因可能是由于生境地人为的破坏,如当地居民肆意的砍伐。在中国的地理分布区,除了在保护区的居群以外,其他居群都受到不同程度的人为砍伐,当地居民用来当柴火使用,而在日本的自然地理分布区,几乎没有被人为破坏的痕迹。
AMOVE分析结果显示,双花木属的变异主要来自中国和日本两个群体间,变异占53.08%,而组内居群间的变异只占24.11%,来自个体间的变异也只有22.80%。该结果与PERMUT计算结果相符,双花木属中国和日本两个群体间遗传分化系数Gst为0.796,且Nst(0.905)> Gst(P<0.01),说明双花木属植物亲缘关系较近的单倍型发生在同样居群中的概率极高,且存在着极显著的谱系地理学结构。与Enstrand和Elam[26]认为Gst>0.1意味居群间分化程度较高的研究结果一致。叶绿体DNA(cpDNA)是母系遗传,因此在探讨造成居群间遗传分化的因素时主要考虑种子的传播。双花木属种子仅靠蒴果开裂时的弹力传播[27],且种子的萌发受自身因素限制和外界条件的干扰较大,该属种子发育不良,空壳多且种皮厚,不利于种子发芽,存在“花多果少”和严重的“大小年”结果现象[28]。另外,该属植物种子萌发对温度、光照及水分等环境因素敏感,自然条件下种子的繁殖率极低[29]。因此,当种子通过风力传播到较远的地方时,由于环境的改变影响了种子的萌发,使得种子成活率较低,导致在两个亚种间缺乏基因交流产生了遗传分化。其次,该属植物两个亚种间存在的地理隔离屏障可能是造成其较高遗传分化水平的另一个原因。虽然在末次冰期,中国东海可能作为连接中国大陆与日本的陆桥,但是双花木属植物对环境的依赖性较强,当第四纪末次冰期气候来临时,暴露的陆桥地区没有适合双花木属生长的环境,因此阻碍了该物种的迁移和传播,致使了两个亚种间的遗传分化。另外,双花木属为较原始的类群,所以在漫长的进化历史过程中,两个地区间的双花木属群体在分子遗传上积累了大量可以有效区分彼此的核苷酸变异,导致两个亚种间的遗传分化。
3.2 系统发育分析通过cpDNA 3个片段的联合分析,基于PAUP和Bayes的系统发育树结果都支持日本的D. Cercidifolius和中国的D. cercidifolius subsp. Longipes为单系类群,且结果进一步支持日本群体分为两个地理组,一个来自日本本州岛的中部(居群QF、CY),一个是来自日本本州岛的西南部(居群gz、gd和JH)。系统发育树的结果显示,日本的6个居群分化较中国的居群明显,日本的6个居群除了gs居群只有单一的一种单倍型外,其他居群都检测到两种或两种以上的单倍型;中国的长柄双花木群体则所有居群都只检测到单一的一种单倍型,且除了官山(GS)和井冈山(JG)有独享单倍型外,其他居群都共享单倍型H1,两个群体间无共享单倍型,因此,尽管两个亚种在形态学上的差异有限,差异有限的原因推测与中国-日本具有相似的地质因素有关,两个地区之间的个体是否能够进行杂交以及杂交成种后的生存和繁育情况有待进一步研究,但是本研究的结果从分子遗传学的角度有力的支持了两个亚种的分类地位。双花木属中-日间断分布格局形成的原因可以用地理隔离假说[30]解释,在地理隔离情况下,两个群体被隔离后,基因流消失,随着突变和遗传漂变的积累,群体间产生分化。而新产生的单倍型不能扩张到被隔离的另一个地区,最终导致群体间无共享单倍型。虽然Harrison等的研究结果表明,在末次冰期,中国与日本地区之间可能被一条狭长连续的落叶林所覆盖,存在着可能的陆桥连接着这两个地区,有望为这两个地区的双花木属群体提供基因交流的机会。但是,本研究的结果表明,双花木属中国和日本的两个群体间无共享单倍型,且产生了较大的遗传分化水平,说明两者在该时期未能出现这个假设的情况。可能的解释为说明该陆桥连接存在局部地区生境片段化现象,阻碍了两个群体之间的基因流。类似的情况已有研究报道,孙逸[31]对东亚特有濒危植物黄山梅属(Kirengeshoma)的亲缘地理学研究结果表明,黄山梅属种下中国和日本两个地区的黄山梅在2.71个百万年开始分化,即上新世晚期(Lower Pliocene),提出上新世中期气候的波动可能是黄山梅群体中-日间断分布的原因,末次冰期存在的可能的陆桥未能给两个群体提供基因交流的机会。祁新帅[30]对东亚间断分布植物蛛网萼(Platycrater arguta Sieb. et Zucc)的生物地理学研究中,提出中国东海的冰期陆桥功能并不广泛适用于陆桥两侧的所有落叶林地植物这一观点,陆桥的存在对中-日间断分布的蛛网萼植物几乎没有起到任何作用,本研究的结果从侧面支持了以上学者的研究结果。
4 结论基于PAUP和Bayes的系统发育树拓扑结构一致,结果都支持日本的D. Cercidifolius和中国的D. cercidifolius subsp. Longipes为单系类群,两个群体间无共享单倍型,缺乏有效的基因交流,且结果进一步支持日本群体分为两个地理组,一个来自日本本州岛的中部(居群QF,CY),一个是来自日本本州岛的西南部(居群gz、gd和JH)。
| [1] | 廖飞勇. 长柄双花木的生理习性及其在园林中的应用[J]. 北方园艺, 2010(8): 69-71. |
| [2] | 沈如江, 林石狮, 凡强, 等. 江西省三清山长柄双花木优势群落研究[J]. 武汉植物学研究, 2009, 27(5): 501-508. |
| [3] | 谢国文, 谭巨清, 曾宇鹏, 等. 国家重点保护物种长柄双花木南岭群落植物区系与资源[J]. 广东教育学院学报, 2010, 30(5): 79-87. |
| [4] | Morawetz W, Samuel MRA. Karyological patterns in the Hamame-lidae//Crane PR, Blackmore S. eds., Evolution, Systematics and Fossil History of the Hamamelidae[M]. Oxford: Clarendon Press, 1987, 1: 129-154. |
| [5] | Oginuma K, Tobe HK. Karyomorphology and evolution in some Ha-mamelidaceae and Platanaceae(Hamamelidae: Hamamelidales)[J]. Bot Mag Tokyo, 1991, 104: 115-135. |
| [6] | 潘开玉, 杨亲二. 双花木属和壳菜果属(金缕梅科)的核型研究[J]. 植物分类学报, 1994, 32(3): 235-239. |
| [7] | Shi SH, Chang HT, Chen YQ, et al. Phylogeny of the Hamamelidaceae based on the ITS sequences of nuclear ribosomal DNA[J]. Biochem Syst Eco, 1998, 26: 55-69. |
| [8] | Li JH, Bogle AL, Klein AS. Phylogenetic relationships in the Hamamelidaceae: evidence from the nucleotide sequences of the plastid gene matK[J]. Plant Syst Evol, 1999, 218: 205-219. |
| [9] | Wen J, Shi SH. A phylogenetic and biogeographic study of Hamamelis(Hamamelidaceae), an eastern Asian and eastern North American disjunct genus[J]. Biochem Syst Eco, 1999, 27: 55-66. |
| [10] | Ickert-Bond SM, Wen J. Phylogeny and biogeography of Altingiaceae- Evidence from combined analysis of five non-coding chloroplast regions[J]. Mol Phylogenet Evol, 2006, 39: 512-528. |
| [11] | 袁长春, 张锡亭. 金缕梅科各亚科间的亲缘关系分析[J]. 湘潭师范学院学报, 1999, 20(3): 97-100. |
| [12] | 侯新东, 尹帅, 盛桂莲, 等. 基于ITS序列探讨10种荨麻科植物的系统发育关系[J]. 生物技术通报, 2013(8): 68-73. |
| [13] | 陈诚, 沈和定, 吴文健, 等. 基于28SrDNA部分序列的石磺科系统发育研究[J]. 生物技术通报, 2010(6): 172-178. |
| [14] | 谢国文, 赵俊杰, 李荣华, 等. 濒危植物长柄双花木ISSR-PCR反应体系的优化[J]. 广州大学学报: 自然科学版, 2010, 9(2): 45-50. |
| [15] | Oxelman B, Liden M, Berglund D. Chloroplast rps16 intron phylogeny of the tribe Sileneae(Caryophyllaceae)[J]. Plant Syst Evol, 1997, 206: 393-410. |
| [16] | Sang T, Crawford D, Stuessy T. Chloroplast DNA phylogeny reticulate evolution and biogeography of Paeonia(Paeoniaceae)[J]. American Journal of Botany, 1997, 84(8): 1120. |
| [17] | Huang YL, Shi SH. Phylogenetics of Lythraceae sensu lato: a preliminary analysis based on chloroplast rbcL gene, psaA-ycf3 spacer, and nuclear rDNA internal transcribed spacer(ITS)sequences[J]. Int J Plant Sci, 2002, 163: 215-225. |
| [18] | Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program or indows 95/98/NT[C]//Nucleic Acids Symposium Series; 1999, 41: 95-98. |
| [19] | Librado P, Rozas J. DNA SP v5: a software for comprehensive analysis of DNA polymorphism data[J]. Bioinformatics, 2009, 25(11): 451-1452. |
| [20] | Excoffier L, Laval G, Schneider S. Arlequin(version 3. 0): an integrated software package for population genetics data analysis[J]. Evolutionary Bioinformatics Online, 2005: 1- 47. |
| [21] | Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure[J]. Evolution, 1984: 1358-1370. |
| [22] | Nei M. Molecular evolutionary genetics[M]. New York: Colum-bia University Press, 1987. |
| [23] | Posada D, Crandall KA. Modeltest: testing the model of DNA substitution[J]. Bioinformatics, 1998, 14(9): 817-818. |
| [24] | Swofford D. PAUP. Phylogenetic analysis using parsimony and other Methods, Version 4. 0b 10[M]. Sunderland(MA, USA): Sinauer Associates, 2003. |
| [25] | Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees[J]. Bioinformatics, 2001, 17(8): 754-755. |
| [26] | Ellstrand NC, Elam DR. Population genetic consequences of small population size; implication for plant conservation[J]. Annual Review of Ecology Systematics, 1993, 24: 217-242. |
| [27] | 高浦新, 李美琼, 周赛霞, 等. 濒危植物长柄双花木(Disanthus cercidifolius var. longipes)的资源分布及濒危现状[J]. 植物科学学报, 2013, 31(1): 34-41. |
| [28] | 史晓华, 徐本美, 黎念林, 等. 长柄双花木种子休眠与萌发的初步研究[J]. 种子, 2002, 6: 5-7. |
| [29] | 肖宜安. 濒危植物长柄双花木繁殖生态学与光适应性研究[D]. 重庆: 西南师范大学生命科学学院, 2005. |
| [30] | 祁新帅. 东亚特有间断分布植物殊网薷厲的生物地理学研究[D]. 杭州: 浙江大学, 2013. |
| [31] | 孙逸. 东亚特有濒危植物黄山梅的亲缘地理学与群体遗传学研究[D]. 杭州: 浙江大学, 2012. |