




2. 中山大学附属第一医院,广州 510080
2. The First Affiliated Hospital of Sun Yat-sen University,Guangzhou 510080
AIP1(Apoptosis signal-regulating kinase 1-interacting protein-1,又称为DAB2IP)是近来鉴定的Ras-GTP酶活化蛋白家族新成员,其基因定位于人染色体9q33.1-9q33.3上,含有1个开放阅读框架,cDNA全长6.3 kb,编码1 065个氨基酸、分子量为120 kD左右的蛋白质。AIP1蛋白的N端有参与膜蛋白定位的PH结构域、细胞信号凋亡调节酶1结合有关C2结构域和抑制Ras信号通路的GAP结构域;C端有抑制核转录因子NF-κB信号通路有关的PER结构域和调控PI3K-Akt信号通路的PR结构域[1]。AIP1是新发现的一种抑癌基因,它通过介导多种肿瘤相关信号通路,如MAPK-ERK、PI3K-Akt、ASKl-JNK和GSK-3β-β-catenin等,影响细胞的增殖、存活和凋亡[2, 3, 4]。研究发现AIP1在前列腺癌、肝细胞癌、肺癌、乳腺癌、胃癌等多种恶性肿瘤的发生发展中起重要作用[5, 6, 7, 8, 9]。
目前,普遍使用基因敲减技术敲低目的细胞AIP1基因研究AIP1基因功能,但是基因敲减技术仅影响mRNA降解,不能完全沉默目的细胞AIP1基因的表达,对充分研究AIP1基因功能有一定的局限性。本实验利用CRISPR/Cas9系统构建AIP1基因敲除质粒,并用慢病毒系统进行包装,旨在建立CRISPR/Cas9慢病毒系统敲除人源性细胞系AIP1基因的方法,为深入研究AIP1基因功能奠定基础。 1 材料与方法 1.1 材料
人胚肾细胞株(293T)购于美国ATCC细胞库,PX458和lentiCRISPRv2质粒购于Addgene公司,BbsⅠ酶、BsmBⅠ酶、T4 DNA连接酶和T7E1酶购自New England Biolabs (NEB)公司,质粒抽提试剂盒购自Qiagen公司,DNA提取试剂盒购自TaKaRa公司,Lipofectamine® 2000试剂、高糖.DMEM培养基、胎牛血清和胰酶均购自Gibco公司,寡核苷酸序列由上海捷瑞公司合成。 1.2 方法 1.2.1 sgRNA寡核苷酸链合成
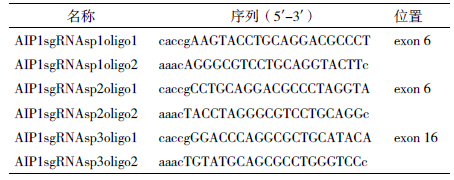
我们使用CRISPR在线设计工具(http://crispr.mit.edu/)根据评分系统,分别在AIP1的外显子6和16设计3个20 bp的sgRNA (sp1、sp2和sp3)(图 1)。分别在编码链模板5'端添加CACCG,非编码链模板5'端添加AAAC,3'端添加C,设计3对CRISPR寡核苷酸链,由上海捷瑞公司合成。设计sgRNA及序列见表 1。
 |
| 图 1 CRISPR/Cas靶序列在AIP1基因的位点及扩增靶序列位点引物示意图 |
将两条sgRNA寡核苷酸单链化学合成双链,连接至BbsⅠ酶切线性化的PX458质粒;转化到stbl3感受态细胞中,挑取阳性单克隆菌落抽提质粒并送广州艾基生物测序验证。将重组质粒分别命名为:PX458-sgRNAsp1、PX458-sgRNAsp2和PX458-sgRNAsp3。将sgRNAsp2与BsmBⅠ酶切后的lentiCRISPRv2载体使用上述方法连接,并测序验证。 1.2.3 敲除效率验证
细胞培养和细胞转染:293T细胞用含10%胎牛血清的高糖DMEM培养基于5% CO2,37℃恒温培养。转染前24 h,取对数期细胞以5×105/孔接种到6孔板培养,将其分组为:PX458(阴性对照组)、PX458-sgRNAsp1、PX458-sgRNAsp2和PX458-sgRNAsp3。将相应的敲除质粒及阴性对照质粒各2 μg经Lipofectamine® 2000试剂瞬时转染到293T细胞中,转染48 h后消化收集各组细胞,提取基因组DNA。采用T7E1实验进一步验证敲除效率:根据BLAST分别设计3条靶向序列的引物(表 2)。PCR扩增目的片段,纯化后使用PCR仪进行变性退火。加入10 U T7E1酶,37℃水浴15 min,加入2 μL 0.5 mol/L EDTA终止反应。使用2%的琼脂糖凝胶DNA电泳,检测敲除效率。

将重组质粒lentiCRISPRv2-sgRNAsp2和lentiCRISPRv2空载体各1.2 μg分别与包装质粒pCMV-R8.2 0.6 μg及辅助质粒VSV-G 1.2 μg混合,分别稳定转染293T细胞;转染12 h后,弃去培养基,加入3 mL培养基置37℃继续培养;48 h后收集,0.45 μm滤器过滤并分装病毒上清液。加入新鲜培养基继续培养;24 h后再次收集含病毒上清液的培养基。-80℃保存。 1.2.5 嘌呤霉素对293T 细胞的最小致死量筛选
将293T细胞种入6孔板培养,待细胞融合度达到70%-80%时,按照下列浓度加入嘌呤霉素(μg/mL):0、0.5、1.0、1.5、2.0和2.5,隔天换液,并保持培养基的嘌呤霉素浓度恒定。第3天能够使293T细胞全部死亡的最小浓度为1.0 μg/mL。 1.2.6 构建稳定细胞株
待293T细胞融合度达到50%时更换1 mL培养基,加入1 mL病毒上清液和1.6 mL polybrene (终浓度8 μg/mL),6 h后换正常培养基培养。48 h后重复感染一次,感染完毕后换正常培养基培养。48 h后加入嘌呤霉素(1.0 μg/mL),维持嘌呤霉素浓度7 d,对于筛选出的阳性克隆,消化收集各孔细胞并计数,采用有限稀释法稀释至终浓度为每100 μL培养基0.5个细胞,按每孔200 μL细胞稀释液加至96孔板[10]。显微镜下观察只含有单个细胞的孔并标记。待单个细胞生长至细胞团时逐级扩大培养,取少量细胞提取基因组DNA,经PCR扩增后测序与野生型基因组对比,检测AIP1是否成功敲除。对缺失突变碱基最多的细胞株进行扩大培养,提取细胞蛋白,Western blot检测单克隆细胞AIP1蛋白表达量。 2 结果 2.1 重组质粒载体的检测结果
分别在AIP1的3条靶序列添加粘性末端,化学合成sgRNAsp1、sgRNAsp2和sgRNAsp3与酶切后的PX458质粒连接,转化到stbl3感受态细胞中,挑取阳性单克隆菌落抽提质粒。广州艾基生物公司测序结果(图 2)显示,sgRNAsp1、sgRNAsp2和sgRNAsp3分别正确插入pX458载体中。
 |
| 图 2 质粒序列测序图 |
将PX458空载体及PX458-sgRNAsp1、PX458-sgRNAsp2和PX458-sgRNAsp3分别瞬时转染至293T细胞,转染48 h后,荧光显微镜下观察4组293T细胞都有绿色荧光蛋白表达(图 3),消化收集各组细胞,提取基因组DNA。PCR扩增含有sgRNA的目的片段,纯化后加入T7E1酶反应,琼脂糖凝胶DNA电泳发现与对照组比较sgRNAsp1、sgRNAsp2和sgRNAsp3都有两条切开的条带,其中sgRNAsp2切开效果最显著,表明AIP1sgRNAsp2靶向敲除效率最高(图 4)。
 |
| 图 3 PX458空载及3组PX458-sgRNA转染至293T细胞 |
 |
| 图 4 检测3个sgRNA的靶向敲除效率 |
为了提高转染效率,将sgRNAsp2连接到lentiCRISPRv2慢病毒质粒,和空载体分别转染至293T细胞,收集病毒液,感染正常293T细胞,嘌呤霉素筛选出阳性克隆,有限稀释法稀释至单个细胞,逐级扩大培养,培养出6个单克隆稳定细胞株与其野生型基因组DNA比较,发现目的片段中出现了不同碱基数的缺失突变和插入突变(图 5)。
 |
| 图 5单克隆细胞在目的sgRNA位点的缺失突变 |
对缺失突变碱基最多的细胞株和转染空载体细胞株进行扩大培养,提取细胞蛋白,Western blot检测敲除AIP1基因的293T细胞与对照组比较,发现AIP1蛋白表达缺失(图 6),表明已敲除393T细胞中AIP1基因。
 |
| 图 6 稳定细胞株中AIP1蛋白的表达 |
随着生物学研究的发展,基因组编辑技术为了解特定基因的功能提供了基础。目前ZFNs和TALENs技术突破了传统的基因组定点编辑,但其具体操作对实验要求较高,耗时较长,较难广泛普及。而CRISPR/Cas9系统可以对任何后面紧随NGG (PAM)的20 bp的序列进行编辑,能同时作用于多个靶位点[11]。此外,载体构建简单,针对特定基因位点只需要设计sgRNA,不需要对每个基因编辑位点都设计和组装两个核酸酶。CRISPR/Cas9系统具有简单灵活、特异性高、细胞毒性低等特点,可以对特定基因组位点进行切割置换[12]。与RNAi技术比较,CRISPR/Cas9系统通过RNA识别DNA序列然后再改变DNA序列是可以遗传的[13]。本研究构建的lentiCRISPRv2-sgRNAsp2质粒携带有化脓性链球菌Ⅱ型CRISPR/Cas9系统的Cas9核酸内切酶基因,在转染进细胞后可以表达该核酸内切酶。通过T7E1实验检测sgRNA靶位点的切割效率,发现sgRNAsp2在AIP1基因目的位点产生切割的效率高达51.8%。连接sgRNAsp2到慢病毒lentiCRISPRv2载体,更高效稳定的转入293T细胞。在细胞亚克隆中,有6个单克隆细胞基因组在目的片段中出现了不同碱基数的突变。对缺失突变碱基最多的细胞株进行扩大培养,建立稳定敲除AIP1的293T细胞株。
将目的基因连接到载体并转染至目的细胞是获得长期稳定表达细胞株的关键步骤。目前,转染目的细胞常用的基因导入载体主要是质粒载体和病毒载体。质粒载体适用于短时间的活细胞示踪,转染率相对较低,且对目的基因干预作用弱。与质粒载体相比,病毒载体具有转染率高、目的基因长期稳定表达及适用范围广等优势。因此,越来越多的研究人员更倾向于使用病毒载体转染,包括腺病毒、慢病毒和逆转录病毒3种表达系统,其中慢病毒载体系统适用细胞类型广泛,可稳定转染目的基因,能整合外源基因进入目的细胞达到稳定表达,是目前基础和临床应用研究中使用最广泛的一类载体。本实验利用慢病毒载体系统转染293T细胞具有较高的转染率,成功的获得长期、稳定敲除AIP1基因的细胞株。
最近研究发现,AIP1作为一个新发现的抑癌基因,不仅在肿瘤细胞的增殖存活和凋亡过程中扮演着重要角色,还参与调控动脉硬化、腹主动脉瘤及早发性心肌梗死等多种非肿瘤性疾病的发生发展[14, 15]。本研究构建目的细胞AIP1基因敲除的细胞系,是深入研究AIP1基因功能的前提基础。 4 结论
本研究结合慢病毒系统的稳定转染功能和CRISPR/Cas9系统的敲除功能,建立了CRISPR/Cas9慢病毒系统,充分发挥了慢病毒包装系统与Cas9系统的优势,永久性敲除目的细胞AIP1基因,获得敲除AIP1基因的稳定细胞株。证实了CRISPR/Cas9慢病毒系统的可行性,为进一步构建其他AIP1基因敲除的人源性细胞系提供了一种简单、高效的方法。
| [1] | Min W,Pober JS.AIP1 in graft arteriosclerosis[J].Trends Cardiovasc Med,2011,21(8):229-233. |
| [2] | Chen H,Pong R,Wang Z,et al.Differential regulation of the human gene DAB2IP in normal and malignant prostatic epithelia:Cloning and characterization[J].Genomics,2002,79(4):573-581. |
| [3] | Min J,Zaslavsky A,Fedele G,et al.An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-κB[J].Nature Medicine,2010,16(3):286-294. |
| [4] | Chen H,Toyooka S,Gazdar AF,et al.Epigenetic regulation of a novel tumor suppressor gene (hDAB2IP) in prostate cancer cell lines[J].J Biol Chem,2003,278(5):3121-3130. |
| [5] | Xie D,Gore C,Liu J,et al.Role of DAB2IP in modulating epithelialto-mesenchymal transition and prostate cancer metastasis[J].Proc Natl Acad Sci USA,2010,107(6):2485-2490. |
| [6] | Qiu G,Xie H,Wheelhouse N,et al.Differential expression of hDAB2IPA and hDAB2IPB in normal tissues and promoter methylation of hDAB2IPA in hepatocellular carcinoma[J].Journal of Hepatology,2007,46(4):655-663. |
| [7] | Yano M,Toyooka S,Tsukuda K,et al.Aberrant promoter methylation of human DAB2 interactive protein (hDAB2IP) gene in lung cancers[J].International Journal of Cancer,2005,113(1):59-66. |
| [8] | Yano M,Toyooka S,Tsukuda K,et al.Aberrant promoter methylation of human DAB2 interactive protein (hDAB2IP) gene in lung cancers[J].Int J Cancer,2005,113(1):59-66. |
| [9] | Dote H,Toyooka S,Tsukuda K,et al.Aberrant promoter methylation in human DAB2 interactive protein (hDAB2IP) gene in gastrointestinal tumour[J].Br J Cancer,2005,92(6):1117-1125. |
| [10] | Harms DW,Quadros RM,Seruggia D,et al.Mouse genome editing using the CRISPR/Cas system[J].Curr Protoc Hum Genet,2014,83:15-17. |
| [11] | Yang H,Wang H,Shivalila CS,et al.One-Step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering[J].Cell,2013,154(6):1370-1379. |
| [12] | Fu Y,Foden JA,Khayter C,et al.High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells[J].Nature Biotechnology,2013,31(9):822-826. |
| [13] | Carroll D.Staying on target with CRISPR-Cas[J].Nat Biotechnol,2013,31(9):807-809. |
| [14] | Gretarsdottir S,Baas AF,Thorleifsson G,et al.Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm[J].Nature Genetics,2010,42(8):692-697. |
| [15] | Yu L,Qin L,Zhang H,et al.AIP1 prevents graft arteriosclerosis by inhibiting interferon-gamma-dependent smooth muscle cell proliferation and intimal expansion[J].Circ Res,2011,109(4):418-427. |