




2. 北京大学生命科学学院,北京 100871
2. School of Life Science,Peking University,Beijing 100871
肝细胞癌(Hepatocellular carcinoma,HCC)是一种致死率很高的恶性肿瘤,2012年全球范围内发病率位居肿瘤第五位,死亡率高居肿瘤第二位[1]。严峻的临床诊治形势对HCC发生及转移机制研究提出迫切需求,HCC相关机制的深入阐释对HCC早期诊断、药靶研发及治疗预后具有重要意义。但HCC致病因素多,病程复杂,涉及通路广,因此研究难度较大,其发生发展机制目前尚不完全清楚。
动物模型作为重要的研究手段,在HCC发生发展机制研究中发挥了不可或缺的作用。化学诱导、原位异位移植及转基因等方法在目前HCC动物模型构建中较为常用。转基因动物模型与其他常用HCC动物模型相比,在研究特殊基因在HCC发生过程中的作用或不同基因间的相互作用,以及与肝脏特异性致癌物之间的关系中具有独特优势,迅速成为新的研究热点。本文将对近年来常用的HCC转基因小鼠模型进行分类介绍,并对其在HCC相关机制研究及药物筛选中的应用进行综述,旨为相关领域研究者提供参考。
1 HCC转基因动物模型概况转基因动物模型是通过基因工程方法导入或敲除动物体内特定基因,从而影响动物性状表达并产生稳定遗传修饰的动物模型。1982年,Gordon等[2]首次利用显微注射法成功构建转基因小鼠,此后转基因动物模型得到快速发展和广泛应用。
HCC转基因模型的实验动物多为CD1、C57BL/6×CBA/J、C57BL/6×DBA2等品系的小鼠。HCC转基因小鼠主要通过重组基因敲入(Knock-in)或目的基因敲除(Knock-out)构建,利用显微注射法将重组基因导入受精卵是构建转基因小鼠的经典方法。另外,通过逆转录病毒介导等方式对小鼠胚胎干细胞进行基因靶向修饰也较常用。外源重组基因构建时通常包含肝脏特异性蛋白启动子元件,可在肝脏进行组织特异性表达。白蛋白(Albumin)启动子是最常用的启动子。此外,α-1-抗胰蛋白酶(α-1-antitrypsin,AAT)、金属硫蛋白(Metallothionein)等启动子也有较多应用。敲入动物体内的外源基因可以为单基因(如HBx、SV40等),也可以为双基因(如c-Myc + TGF-α、β-Catenin + H-ras等),单基因敲入成功率较双基因的成功率高,但发生HCC所需时间较长。
根据所修饰基因的不同,HCC转基因小鼠模型可主要分为病毒基因转基因模型、原癌基因转基因模型、生长因子及肿瘤微环境相关基因转基因模型等,具体信息参见表 1。
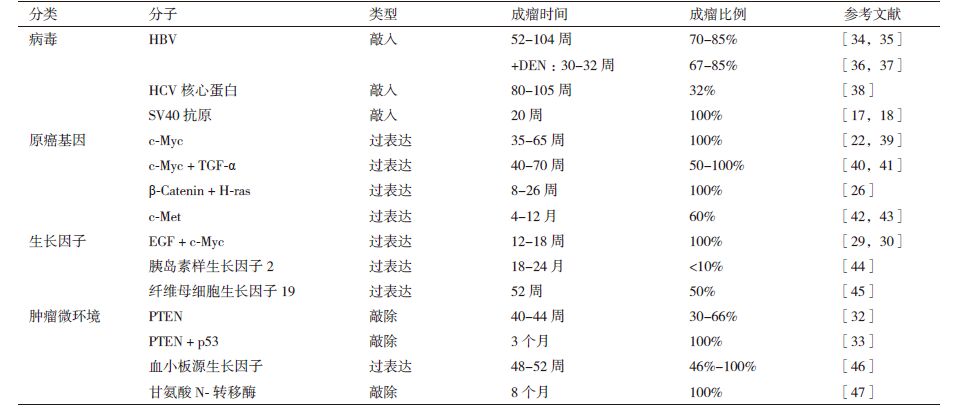
病毒基因转基因模型所采用的基因主要有乙型肝炎病毒(Hepatitis B virus,HBV)和丙型肝炎病毒(Hepatitis C viral,HCV)等肝炎病毒基因及猴空泡病毒(Simian vacuolating virus,SV40)等,其中肝炎病毒转基因模型是目前最常用的模型,SV40病毒近年也受到较多关注,成为常用的建模方法之一。
2.1 HBV转基因模型HBV感染是全球范围内HCC的主要诱因之一。HBV在长期感染宿主的情况下,病毒基因可整合至宿主基因组中,导致基因突变、染色体不稳定及基因组重排,从而引发HCC。HBV病毒基因组包括4个开放阅读框,分别为编码包膜蛋白的S基因(Surface)、编码e抗原和核心抗原的C基因(Core)、编码聚合酶的P基因(Polymerase)以及编码HBx蛋白的X基因。
X基因是目前构建HBV转基因HCC模型时采用最多的基因,其编码产物HBx蛋白是一个反式转录激活因子,可促进一系列原癌基因的表达,诱导HCC产生。Kim等[3]利用显微注射法将X基因及其启动子和增强子转入小鼠受精卵中,成功构建HBx转基因小鼠,是目前应用较多的HCC模型之一。目前研究中,HBx转基因小鼠HCC发生于44-140周左右,成瘤率约为70%-85%。HBx模型在HBV诱发HCC机制研究中应用广泛,揭示了多种HBx促肝癌发生的机制,如参与DNA修复及合成[4]、影响细胞周期[5]以及加速肝细胞凋亡[6]等。近年来应用HBx模型也得到较多研究成果,Yu等[7]发现病毒-宿主信号通路中的细胞周期相关激酶(Cell cycle-related kinase,CCRK)可能在HBV诱发HCC过程中起到重要作用,同时,CCRK还是雄激素受体通路中的关键蛋白,可能是导致HCC的男性主导性别差异的原因之一;Quétier等[8]通过对实施部分肝切除术的HBx转基因小鼠进行研究发现,HBx蛋白可通过上调白介素-6(IL-6)的表达影响部分肝切除术后的肝细胞增殖,说明HBx可能参与到肝脏疾病的细胞周期失调过程中。另外,HBx模型在HCC药物研究方面也有重要应用,在抗癌药物如水飞蓟素(Silymarin)[9]、β-干扰素(Interferon-β)[10]等对HBV相关HCC的药理作用研究中起到关键作用。
HBV基因中编码表面抗原(HBsAg)的S基因同样能用于HCC模型的构建,但HBsAg转基因小鼠模型表现出的炎症及HCC类型与HBx转基因模型差异较大。Chisari等[11]构建的过表达HBsAg的转基因小鼠目前最为常用,模型在4个月左右表现出慢性肝损伤的特征,逐渐表现出不同程度的炎症、肝再生、转录失调等症状,18个月左右出现肉眼可见的肿瘤。利用此模型有助于研究肝细胞损伤与HCC发生之间的关系,证明持续、严重的肝细胞损伤会引起一系列增殖反应,导致肝细胞生长失控,HCC发生率增加。
2.2 HCV转基因模型HCV感染是诱发HCC的重要因素之一,慢性HCV感染会引起肝硬化,并增加HCC发生的风险。HCV基因组是一个正义单链RNA分子,由一个开放阅读框组成,可编码一个多聚糖蛋白,经修饰后形成10个病毒活性蛋白。
HCV核心蛋白可通过影响脂类和甘油酸三酯间的转换,以及影响代谢相关转录因子活性,促进肝细胞脂肪变性和氧化应激。Koike和Kamegaya等[12, 13]通过显微注射方法,利用HCV核心蛋白、包被蛋白E1和E2及HCV全长蛋白基因分别建立转基因小鼠模型发现,只有在HCV核心蛋白存在时模型小鼠才能发展出HCC,包被蛋白E1-E2转基因小鼠中无HCC发生。第16个月时,32%的HCV核心蛋白模型雄鼠可发生HCC,而HCV全长蛋白模型(核心蛋白-包被蛋白E1-E2)可在13个月时表现出腺癌和HCC症状,这为HCV蛋白可直接致癌提供了有力证据。在HCV致癌机制研究中,Benzoubir等[14]通过应用HCV核心蛋白转基因小鼠发现HCV可能通过激活转化生长因子-β(Transforming Growth Factor-β,TGF-β)调节肝细胞内相关信号通路及影响基质微环境,从而诱发肝癌;Fujinaga等[15]发现HCV可引起活性氧(Reactive oxygen species)的过度产生并抑制肝内的抗氧化系统,引起氧化应激,加速HCC的发生。
2.3 SV40抗原转基因模型SV40是一种DNA病毒,其基因组编码的大T抗原和小t抗原具有致癌功能,主要通过结合并灭活宿主细胞p53、Rb等抑癌基因,促进DNA复制,从而引发癌症。
SV40 T-抗原转基因小鼠主要通过显微注射方法将具有肝脏特异性蛋白启动子的重组基因导入受精卵构建。另外,也有研究者利用Cre/LoxP系统成功构建肝脏特异表达SV40 T-抗原的转基因小鼠[16]。SV40 T-抗原转基因小鼠模型建立后,模型在很短的时间(4-12周)内即可发展出HCC,并且能发生肺转移[17, 18]。SV40抗原转基因小鼠多用于研究HCC发生发展中免疫活动的变化,Willimsky 等[19]构建SV40大T抗原(TAg)转基因模型发现,模型小鼠在早期HCC阶段T细胞侵润程度远高于晚期HCC阶段,说明进展的局部免疫抑制阻碍了T细胞侵润,并发现程序性细胞死亡蛋白(Programmed Cell Death Protein-1,PD-1)及其配体(PDL-1)在抗原特异性局部耐受中发挥了重要作用。此外,SV40转基因模型也为多种基因如NM23[20]、FZD7[21]等在HCC发生发展中的作用机制研究提供了重要平台。
3 原癌基因转基因模型原癌基因可编码参与肿瘤发生发展过程的蛋白质,其激活或过表达会大大增加肿瘤发生的概率。目前在转基因小鼠中研究较多的过表达原癌基因产物有c-Myc蛋白和β-连环蛋白(β-Catenin)等。
3.1 c-Myc转基因模型c-Myc蛋白作为一种转录因子,在细胞增殖、凋亡、分化等过程中发挥重要作用,与包括HCC在内的多种肿瘤密切相关。通过突变或过表达c-Myc蛋白构建HCC小鼠模型可用于研究HCC多步骤发展过程。
过表达c-Myc的转基因小鼠在15周时产生多核型细胞,60%的细胞出现发育异常,60-90周后91%的小鼠发生腺癌,54%为HCC。c-Myc基因与其他原癌基因或生长因子的协同作用在肿瘤发展中发挥关键作用。因此c-Myc转基因小鼠在构建时多与其他基因进行联合过表达,可使建模速度大大提升。双基因模型多采用单基因转基因小鼠杂交筛选的方法获得。c-Myc与TGF-α双基因敲入在4个月时即可使70%的模型小鼠产生异常发育结节,其中18%为HCC[22, 23];c-Myc与E1F2同时肝脏特异过表达的小鼠模型发生HCC的速度较二者分别过表达的小鼠模型有明显提升[24];c-Myc与星形胶质细胞上调基因1(Astrocyte elevated gene-1,AGE-1)在人HCC中均表达上调,通过c-Myc及AGE-1双基因模型发现二者在促进HCC发生发展过程中发挥了协同作用[25]。
3.2 β-Catenin转基因模型β-Catenin是一种具有调节基因转录和细胞间黏附等多种重要功能的转录因子,在肝脏发育和再生中扮演重要角色,其突变或过表达与HCC、肺癌、乳腺癌等多种肿瘤的发生密切相关。β-Catenin的突变可能在HCC发生发展中起作用,但是只过表达或突变β-Catenin的转基因动物模型只能引发肝肿大,不能诱导HCC发生。因此,动物模型中通过β-Catenin诱导HCC需要其它基因的协同。
β-Catenin和H-ras基因的双导入即可在100%小鼠中诱发HCC,早期HCC发生于8周内,约26周后可发展成进展期HCC[26];Stauffer等[27]通过将β-Catenin与AKT(CAT/AKT)共激活构建转基因小鼠模型发现,小鼠在4周内可产生肿瘤灶,通过对CAT/AKT肿瘤的关键分子网络进行分析发现,与脂肪变性和脂质代谢相关的通路发生变化,说明β-Catenin和AKT相关通路在肝细胞中的激活与脂肪生成相关HCC的发生密切相关;Dong等[28]利用β-Catenin与组成型雄烷受体(Constitutive androstane receptor,CAR)构建双基因模型小鼠发现,β-Catenin在肝脏中的完全激活会诱导细胞发生衰老和生长停滞,而当CAR同时激活时,虽然肝脏仍具有正常功能,但却出现细胞增殖失控,肝肿大及致死等现象;CAR激活和β-Catenin的部分激活可诱导肿瘤发生,并且这些肿瘤细胞与β-Catenin阳性的人类HCC具有相似的保守基因表达。该研究表明,CAR和β-Catenin的共同激活可诱导HCC发生,并且定义了HCC小鼠模型与人类HCC的直接关系。
4 生长因子及肿瘤微环境相关转基因模型生长因子在肝细胞的生长调控中发挥重要作用,其过表达可造成肝细胞生长失控,继而导致HCC发生。肿瘤微环境是肿瘤形成的重要影响因素,通过修饰与肿瘤微环境形成相关的基因建立转基因小鼠也是一种重要的建模方法。相关基因研究较多的有表皮生长因子(Epidermal growth factor,EGF)、磷酸脂酶与张力蛋白同源物(Phosphatase and tensin homolog deleted on chromosome ten,PTEN)等。
4.1 EGF转基因模型EGF是最早发现的生长因子,通过与细胞表面表皮生长因子受体结合,启动多种胞内信号通路,促进DNA合成,在细胞生长、增殖及分化过程中起到关键作用。分泌性EGF(lgEGF)的过表达可在6-9月内可引发恶性肝细胞癌,模型在30周左右可因HCC致死[29]。EGF与c-Myc的双基因过表达可加速HCC的发展,在17周左右即引起模型动物的死亡[30]。Gazzana等[31]利用2-DE-MALDI-MS技术对EGF转基因小鼠血清进行蛋白质组学分析,通过与健康小鼠血清对比发现25个表达发生变化的蛋白,其中免疫球蛋白(Immunoglobulins)在HCC小鼠血清中显著下调,而淀粉样P成分(Amyloid component P)及载脂蛋白M(Apolipoprotein M)则显著上调,这为EGF致HCC发生的机制研究提供重要线索。
4.2 PTEN转基因模型PTEN蛋白可调控PKB/AKT通路,参与细胞周期和凋亡调控。PTEN在多种肿瘤中处于突变状态,缺失可导致细胞过度增殖,抑制凋亡,是导致肿瘤发生的重要因素之一。肝脏特异性敲除PTEN的小鼠会表现出肝脏脂肪变性、炎症、纤维化及肿瘤等症状,这与人非酒精性脂肪肝(Non-alcoholic steatohepatitis)的症状非常相似。PTEN缺失引发的HCC在40-44周时可在66%的雄鼠和30%的雌鼠中产生[32],而利用最新的CRISPR/Cas9系统将PTEN及p53同时敲除时,100%的小鼠可在3个月后引发HCC[33]。
5 小结目前转基因HCC动物模型构建体系日趋成熟,在HCC的肿瘤生物学研究中得到广泛应用。但同时,转基因HCC模型仍存在诸如造模成本高、成瘤率不均一、不能完全模拟人类HCC发生发展过程等缺点。人类HCC具有转移复发率高、性别差异明显等特点,这些特点在动物模型中的重现以及相关研究目前也有众多讨论[48, 49],但其针对性的HCC模型仍需进一步的开发和优化。因此,建立与人原发性HCC病程接近、诱癌率高、重复性好的HCC转基因动物模型是今后的重要发展方向之一。
可以预见,转基因模型因其在HCC机制研究中的巨大潜力,会在相当长一段时间内得到持续关注。新的基因改造技术,如CRISPR/Cas9系统的出现,为转基因模型的构建提供了更加有力的工具。结合使用基因打靶与核酸干扰等技术,解决动物转基因效率低和外源基因表达异常的问题,进一步优化目前已有的模型,也是未来HCC转基因动物模型的发展方向。这就要求研究者开发和利用更新的基因技术及其它科学手段,研发出更加符合实验要求,更便于进行各种分析的动物品种。除了不断改进和完善HCC动物模型之外,还需充分利用各种模型,与细胞生物学、分子生物学和多组学等研究相结合,进行从分子到表型、从局部到整体的研究,从而使其更好地服务于HCC的基础理论和应用研究。
| [1] | Stewart BW, Wild CP. World cancer report 2014[C]. International Agency for Research on Cancer, 2014. |
| [2] | Gordon JW, Ruddle FH. Germ line transmission in transgenic mice[J]. Progress in Clinical and Biological Research, 1981, 85:111-124. |
| [3] | Koike K, Moriya K, Iino S, et al. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice[J]. Hepatology, 1994, 19(4):810-819. |
| [4] | 吴一迁, 仇效坤. HBVx 转基因小鼠肝组织基因表达谱的微阵列研究[J]. 肿瘤, 2001, 21(4):235-238. |
| [5] | Madden CR, Finegold MJ, Slagle BL. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions[J]. J Virol, 2001, 75(8):3851-3858. |
| [6] | Pollicino T, Terradillos O, Lecoeur H, et al. Pro-apoptotic effect of the hepatitis B virus X gene[J]. Biomed Pharmacother, 1998, 52(9):363-368. |
| [7] | Yu Z, Gao YQ, Feng H, et al. Cell cycle-related kinase mediates viral-host signalling to promote hepatitis B virus-associated hepatocarcinogenesis[J]. Gut, 2014, 63(11):1793-1804. |
| [8] | Quétier I, Brezillon N, Duriez M, et al. Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice[J]. J Hepatol, 2013, 59(2):285-291. |
| [9] | Wu YF, Fu SL, Kao CH, et al. Chemopreventive effect of silymarin on liver pathology in HBV X protein transgenic mice[J]. Cancer Res, 2008, 68(6):2033-2042. |
| [10] | Yamazaki K, Suzuki K, Ohkoshi S, et al. Temporal treatment with interferon-β prevents hepatocellular carcinoma in hepatitis B virus X gene transgenic mice[J]. J Hepatol, 2008, 48(2):255-265. |
| [11] | Chisari FV, Klopchin K, Moriyama T, et al. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice[J]. Cell, 1989, 59(6):1145-1156. |
| [12] | Koike K, Moriya K, Kimura S. Role of hepatitis C virus in the development of hepatocellular carcinoma:transgenic approach to viral hepatocarcinogenesis[J]. J Gastroenterol Hepatol, 2002, 17(4):394-400. |
| [13] | Kamegaya Y, Hiasa Y, Zukerberg L, et al. Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis[J]. Hepatology, 2005, 41(3):660-667. |
| [14] | Benzoubir N, Lejamtel C, Battaglia S, et al. HCV core-mediated activation of latent TGF-β via thrombospondin drives the crosstalk between hepatocytes and stromal environment[J]. J Hepatol, 2013, 59(6):1160-1168. |
| [15] | Fujinaga H, Tsutsumi T, Yotsuyanagi H, et al. Hepatocarcinogenesis in hepatitis C:HCV shrewdly exacerbates oxidative stress by modulating both production and scavenging of reactive oxygen species[J]. Oncology, 2011, 81(Suppl. 1):11-17. |
| [16] | Lou D Q, Molina T, Bennoun M, et al. Conditional hepatocarcinoge-nesis in mice expressing SV 40 early sequences[J]. Cancer Lett, 2005, 229(1):107-114. |
| [17] | Colvin EK, Weir C, Ikin RJ, et al. SV40 TAg mouse models of cancer[C]. Seminars in Cell & Developmental Biology. Academic Press, 2014, 27:61-73. |
| [18] | Runge A, Hu J, Wieland M, et al. An inducible hepatocellular carcinoma model for preclinical evaluation of anti-angiogenic therapy in adult mice[J]. Cancer Res, 2014, 74(15):4157-4169. |
| [19] | Willimsky G, Schmidt K, Loddenkemper C, et al. Virus-induced hepatocellular carcinomas cause antigen-specific local tolerance[J]. J Clin Invest, 2013, 123(3):1032-1043. |
| [20] | Boissan M, Wendum D, Arnaud-Dabernat S, et al. Increased lung metastasis in transgenic NM23-Null/SV40 mice with hepatocellular carcinoma[J]. J Natl Cancer Inst, 2005, 97(11):836-845. |
| [21] | Nambotin SB, Lefrancois L, Sainsily X, et al. Pharmacological inhibition of Frizzled-7 displays anti-tumor properties in hepatoce-llular carcinoma[J]. J Hepatol, 2011, 54(2):288-299. |
| [22] | Lee JS, Chu IS, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer[J]. Nat Genet, 2004, 36(12):1306-1311. |
| [23] | Santoni-Rugiu E, Nagy P, Jensen MR, et al. Evolution of neoplastic development in the liver of transgenic mice co-expressing c-myc and transforming growth factor-alpha[J]. Am J Pathol, 1996, 149(2):407-428. |
| [24] | Conner EA, Lemmer ER, Sánchez A, et al. E2F1 blocks and c-Myc accelerates hepatic ploidy in transgenic mouse models[J]. Biochem Biophys Res Commun, 2003, 302(1):114-120. |
| [25] | Srivastava J, Siddiq A, Gredler R, et al. Astrocyte elevated gene-1(AEG-1)and c-Myc cooperate to promote hepatocarcinogenesis[J]. Hepatology, 2015, 61(3):915-929. |
| [26] | Harada N, Oshima H, Katoh M, et al. Hepatocarcinogenesis in mice with β-catenin and Ha-ras gene mutations[J]. Cancer Res, 2004, 64(1):48-54. |
| [27] | Stauffer JK, Scarzello AJ, Andersen JB, et al. Coactivation of AKT and β-catenin in mice rapidly induces formation of lipogenic liver tumors[J]. Cancer Res, 2011, 71(7):2718-2727. |
| [28] | Dong B, Lee JS, Park YY, et al. Activating CAR and β-catenin induces uncontrolled liver growth and tumorigenesis[J]. Nat Commun, 2015, 6:5944. |
| [29] | Borlak J, Meier T, Halter R, et al. Epidermal growth factor-induced hepatocellular carcinoma:gene expression profiles in precursor lesions, early stage and solitary tumors[J]. Oncogene, 2005, 24(11):1809-1819. |
| [30] | Liedtke C, Zschemisch NH, Cohrs A, et al. Silencing of caspase-8 in murine hepatocellular carcinomas is mediated via methylation of an essential promoter element[J]. Gastroenterology, 2005, 129(5):1602-1615. |
| [31] | Gazzana G, Borlak J. Mapping of the serum proteome of hepatocellular carcinoma induced by targeted overexpression of epidermal growth factor to liver cells of transgenic mice[J]. J Proteome Res, 2008, 7(3):928-937. |
| [32] | Watanabe S, Horie Y, Kataoka E, et al. Non-alcoholic steatohepatitis and hepatocellular carcinoma:Lessons from hepatocyte-specific phosphatase and tensin homolog(PTEN)-deficient mice[J]. J Gastroenterol Hepatol, 2007, 22(s1):S96-S100. |
| [33] | Xue W, Chen S, Yin H, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver[J]. Nature, 2014, 514(7522):380-384. |
| [34] | Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research[J]. Int J Exp Pathol, 2009, 90(4):367-386. |
| [35] | Koo JS, Seong JK, Park C, et al. Large liver cell dysplasia in hepatitis B virus X transgenic mouse liver and human chronic hepatitis B virus-infected liver[J]. Intervirology, 2005, 48(1):16-22. |
| [36] | Zheng YY, Chen WL, Louie SG, et al. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice[J]. Hepatology, 2007, 45(1):16-21. |
| [37] | Lau CC, Sun T, Ching AKK, et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression[J]. Cancer Cell, 2014, 25(3):335-349. |
| [38] | Tanaka N, Moriya K, Kiyosawa K, et al. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor α in transgenic mice:Implications for HCV-associated hepatocarcinogenesis[J]. Int J Cancer, 2008, 122(1):124-131. |
| [39] | Nevzorova YA, Hu W, Cubero FJ, et al. Overexpression of c-myc in hepatocytes promotes activation of hepatic stellate cells and facilitates the onset of liver fibrosis[J]. Biochim Biophys Acta, 2013, 1832(10):1765-1775. |
| [40] | Griffitts J, Tesiram Y, Reid GE, et al. In vivo MRS assessment of altered fatty acyl unsaturation in liver tumor formation of a TGFα/c-myc transgenic mouse model[J]. J Lipid Res, 2009, 50(4):611-622. |
| [41] | Coulouarn C, Factor VM, Conner EA, et al. Genomic modeling of tumor onset and progression in a mouse model of aggressive human liver cancer[J]. Carcinogenesis, 2011, 32(10):1434-1440. |
| [42] | Tward AD, Jones KD, Yant S, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver[J]. Proc Natl Acad Sci USA, 2007, 104(37):14771-14776. |
| [43] | Wang R, Ferrell LD, Faouzi S, et al. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice[J]. J Cell Biol, 2001, 153(5):1023-1034. |
| [44] | Rogler CE, Yang D, Rossetti L, et al. Altered body composition and increased frequency of diverse malignancies in insulin-like growth factor-II transgenic mice[J]. J Biol Chem, 1994, 269(19):13779-13784. |
| [45] | Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma:ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice[J]. Am J Pathol, 2002, 160(6):2295-2307. |
| [46] | Okada H, Honda M, Campbell JS, et al. Acyclic retinoid targets platelet-derived growth factor signaling in the prevention of hepatic fibrosis and hepatocellular carcinoma development[J]. Cancer Res, 2012, 72(17):4459-4471. |
| [47] | Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice[J]. Hepatology, 2008, 47(4):1191-1199. |
| [48] | Li Y, Tang ZY, Hou JX. Hepatocellular carcinoma:insight from animal models[J]. Nat Rev Gastroenterol Hepatol, 2012, 9(1):32-43. |
| [49] | Bakiri L, Wagner EF. Mouse models for liver cancer[J]. Mol Oncol, 2013, 7(2):206-223. |