




分类是人类认识自然、探索事物本质及其规律的基本出发点之一。对生命形式最早的分类系统能上朔到古希腊哲学家亚里士多德(Aristotle)。对于物种间亲缘关系与分类系统的研究,不仅加深人们对于自然界的认识,还能为一些与人类生命健康有关的应用科学,如医学微生物学与环境元基因组学等带来重要的信息,从而改善人类的生产、生活。传统的物种分类与亲缘关系的研究,无论是林奈(Carolus Linnaeus)的分类系统,还是达尔文(Charles Robert Darwin)在《物种起源》中初次设想的来自共同祖先的亲缘关系,都是根据生物的形态特征。这在动、植物等宏观生物的分类中取得了比较合理的结果。然而占地球上生命物质一半以上的原核生物[1],却很难使用这些传统的方法来实现分类。这是因为当尺度下降到微米级,不但生物形态难以刻画,并且相同的形态可能来自完全不同的物种。1985年Carl Woese和同事们分析当时仅有的约400条16S rRNA序列,提出了基于系统发生的主要细菌门类描述[2]。时至今日,基于16S rRNA序列分析的构树方法现在已经被大多数生物学家接受。尽管16S rRNA序列分析取得了很大成功,但是它在种以下的层次缺乏分辨能力[3, 4]。而环境元基因组学、医药微生物学等应用,区分亚种(Subspecies)、生态型(Ecotypes)、血清型(Serotypes)、生物变种(Biovars)等菌株种以下的分类需求却与日俱增。
全基因组包含了生物全部的遗传信息,其内涵远远丰富于16S rRNA序列,所以分析全基因组数据能够更加准确与细致地研究物种的演化与分类。要提高分辨能力,就需要更好地利用全基因组的信息[5]。而且,随着测序技术的发展,可供研究的全基因组数据越来越多。根据美国国立生物技术信息中心(NCBI)数据显示,截至2015年10月,已完成测序的基因组项目有7 435个,而正在进行的测序计划则多达32 976个,而且这些数字还在快速增加。另外,针对某些特定问题,还有更为庞大的测序计划。例如,2012年8月,美国食品与药物管理局、加州大学戴维斯分校以及安捷伦科技公司发起了旨在对10万种食源性致病菌全基因组的测序计划。针对物种演化研究与分类学的全基因组测序计划,2007年5月,美国能源部联合基因组中心JGI推出“细菌和古细菌基因组百科全书(GEBA)”计划[6];2009年8月,我国深圳华大基因研究院倡导了“万种微生物基因计划”。这些海量的全基因组数据,为基于基因组的系统发生学与分类学的研究提供了丰富的素材。
素材的积累为研究奠定了基础,同时也对研究方法提出了更高的要求。迄今对生物演化与分类的研究,主要基于对单个或少数“同源基因”的序列联配(Sequence alignment)。随着基因组数目增加,这些方法变得不太适用。首先是基因组多样性。就已测序的原核生物基因组而言,即使除去一些高度退化的细菌内共生菌,小的基因组不到50万核苷酸和500个基因[7],而较大的细菌基因组则超过1 300万核苷酸和9 380个基因[8]。这使得挑选“同源基因”变得困难。物种数的增多又使得序列联配遇到计算瓶颈;而且,挑选“同源基因”的做法也不能最大限度地利用基因组信息。因此急需发展不仅不依靠序列联配,同时还能最大限度地利用全基因组信息的研究方法。
我们研究组于2003年提出了基于全基因组的亲缘关系与分类研究方法——组分矢量构树法(Composition vector tree,简称CVTree)[9]。它不需要挑选同源基因,不进行序列比对,从根本上避开了人为干预对结果可能造成的影响。除某些极端情况外,分类结果几乎不受基因组大小的影响,从而非常适合用来构造跨门、跨界,甚至跨超界的生命之树的构建。目前CVTree方法已经成功应用到许多物种的分类研究之中,包括病毒[10]、原核生物[11, 12, 13, 14, 15]、真菌[16]、叶绿体序列[17]及人类的肠道元基因组[18]。研究表明,CVTree具有比传统方法更高的分辨力,这使得CVTree方法有望解决过去难以区分的属内、种内的亲缘关系问题。为了方便用户使用CVTree方法,我们同时开发了网络服务器[19, 20]。为了适应当前基因组数据的海量增加,又开发新版的CVTree网络服务器——CVTree3[21]。除了性能的提高之外,CVTree3服务器还将由CVTree方法生成的亲缘关系树与物种的分类系统自动进行比较,并在网页上以可交互作用的形式显示,为进一步研究原核生物的亲缘关系与分类系统提供方便。本文将简要介绍CVTree核心算法与CVTree3的使用流程,并利用三个典型实例来介绍CVTree3的可能应用。
1 组分矢量方法 1.1 算法——组分矢量构树法组分矢量构树法(CVTree)是一种基于全基因组研究物种亲缘关系的方法。它首先统计基因组中特定长度短串组,为每个物种构造一个高维代表矢量;然后用矢量之间的夹角余弦计算物种间的遗传距离;最后使用邻接法(Neighbor-joining)[22, 23]进行构树。它不需要挑选同源基因,不进行序列联配,从根本上避开了人为干预对结果可能造成的影响。实践表明,基于蛋白质序列的组分矢量方法与传统的分类系统能更好的吻合,下面就以蛋白质序列为例来简要说明CVTree算法。
假设我们需要对一个给定物种,构造基于长度的组分矢量。首先对该基因组的各个基因以长度为窗口,每次滑动一个残基的方式从前向后移动,并求出各种串的出现频度即次数,记为f(a1a2…aK)。则该串的出现概率是:
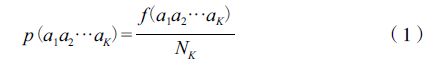
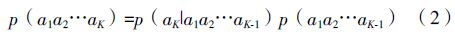
此时做一个Markov假定,假设中K串的出现概率p(a1a2…aK)不依赖于第一个字母a1,则:
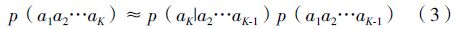
而对于条件概率p(aK|a2a3…aK-1),我们可以通过统计更短的串获得,即:
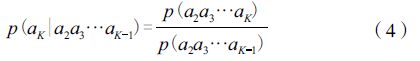
将每类K串对应的考察值v(a1a2…aK) 作为分量构成一个组分矢量。显然当估计值f 0=0时,真值f也为0,此时该维度上的分量设为0。所有这些分量按照统一的固定顺序排列,就得到该物种的组分矢量V=(v1,v2,…,vm) ,其中M=20K。
对于N个物种得到N个这样的组分矢量Vt,其中是物种的编号,介于1与N之间。它们的遗传距离矩阵D是一个对角元素为0的N×N对称矩阵。每个元素对应物种间的遗传距离,由组分矢量的夹角的余弦值给出,其数学表达如下:

最后,基于该遗传距离矩阵D,使用邻接法就可以构建亲缘关系树。
1.2 功能实现——CVTree3网络服务器 1.2.1 基本功能虽然CVTree的算法本身并不复杂,但是要从头实现却也并不容易,所以我们开发了CVTree方法的网络服务器,用户可以通过互联网方便的使用该算法。为了适应当前基因组数据的海量增加,我们开发了最新的CVTree网络服务器CVTree3,用户可以通过 http://tlife.fudan.edu.cn/cvtree3 访问。相对于之前的两个版本的CVTree网络服务器,CVTree3的性能显著提升。就硬件而言,它专享两台具有四路32核、512 G内存的高性能计算集群。同时,为了充分利用计算集群的性能,我们重新设计和编写了核心程序,实现并行化,还从整体上优化了运算过程,从而使效率最大化。此外,为了方便用户在本地使用CVTree方法进行研究,我们还将CVTree3的核心程序单独抽离出来,做成开源的软件包。用户可以从https://www.github.com/ghzuo/cvtree 下载和编译本地版本的CVTree程序。
进入上述地址即可打开CVTree3首页。我们提供了一个Example项目,用户可以在首页点击“Example”按键,查看和浏览这个项目,或者选择“Load/Create Project”新建一个自己项目。点击之后即可得到如图 1所示的项目设置页面。对于每个新建的项目,系统自动分配一个由数字与下划线组成的项目号并且显示在页面顶部(图 1),若从“Example”按键进入,则项目显示为 “example”。在此页面,用户可以选择CVTree方法的基本参数,如:使用DNA序列还是蛋白质序列、短串的长度K(可多选)等。在CVTree3服务器中,我们内置了大量已知分类信息的全基因组,目前包含338种古菌,2 850种细菌,以及8个真核生物作为外类群备选。用户在此页面中部可按类别选择它们,若想逐条选择则点击“See Detail”进入逐条选择页面。此外,用户还可以上传自己的基因组到CVTree3服务器,它们显示在页面的下部。所有参数与基因组都设置好之后,点击右边的绿色按钮“All parameters are fine,Run Project”,即可以提交程序到服务器上运行了。

|
| 顶部数字20151008_2336_24300 为运行项目号,下面依次是状态栏、基本参数设置、内建数据库选择与上传基因组数据 图 1 CVTree3 网络服务器的设置界面顶 |
用户不能修改Example项目,若是由“Example”项目进入该页面,点击该按键则不会有反应。若只使用内建的基因组数据,系统会很快给出结果;若上传了自己的数据,则需要等待。等待时间视上传基因组与选择的内建基因组数目以及相关参数而定。项目在服务器上运行时,用户可以关闭浏览器,这不会影响项目的运行。需要查看项目运行情况与计算结果时,只要使用项目编号从首页导入该项目即可。用户也可以选择在基本参数选项中填入Email,则当项目计算完毕后系统会通知用户。需要注意的是,每个项目在完成后,只在服务器上保持7 d,7 d后系统会自动删除。
1.2.2 亲缘关系与分类的自动比对系统除了性能的提升,将生成的亲缘关系树与分类系统进行自动比较是CVTree3的又一个亮点。服务器在运行的过程中,除了使用CVTee算法进行亲缘关系的分析以外,还会同时得到的亲缘关系与分类系统进行比较。我们综合参考了NCBI分类数据库与Bergey’s Manual等数据,对内建的菌株给出了谱系信息。用户上传的数据,缺省的信息被设置为未知(Unclassified)。用户可以结合已知的谱系信息来推测未知的分类信息,这也是CVTree3网络服务器的一项重要功能。另外,用户可以在上传基因组的方框中上传分类信息文件,上传该信息文件的格式与方法请见用户手册。
所有的计算都进行完毕后,图 1右上角会显示蓝色按钮“See Result”,可以查看计算结果。首先看到的是一个按照菌株的分类阶梯显示的亲缘关系与分类系统的对比结果(图 2)。在对比亲缘关系与分类系统的过程中,我们依靠的关键概念是“单源枝(Monophyly)”。所谓单源枝是指,若某个分类单元刚好对应着亲缘关系树上的一个枝,即分类单元内包含的菌株对应于该分枝下的所有枝叶所代表的菌株。所以,当某个枝为单源时,则说明亲缘关系与分类系统对于当前的数据集合是一致的。除了按分类系统显示单源性,我们还按分类级别统计了单源枝的数目,分别列在图 2的两个未显示的Tab页面“Monophyly”与“None”中。图 2的第4个Tab则列出了一些分类关系不确定即Unclassified菌株,这些菌株在统计过程中并没有被计入。

|
| 图 2 按分类阶梯显示的单源枝截图 |
交互式的亲缘关系树显示是CVTree3的第3个亮点。点击图 2右上角的按钮“See Tree”即可以看到这棵亲缘关系树。图 3是一幅CVTree3亲缘树的截图,用户可以从CVTree3的Example中获得这棵亲缘树,在Web页面上它是一棵动态的树,每个节点都可以打开或收缩,从而调整树的显示方式。此外,与普通的亲缘关系树不同,该亲缘关系树在计算过程中,已自动与分类系统进行了比较与标记,所以用户可以方便的从亲缘树查看每个枝的分类属性。如图 3所示,我们将树展开到门的级别,其中的颜色表明,由CVTree方法得到亲缘树在门的级别上绝大部分与传统的分类系统保持一致。除此之外,我们还提供了很多方便的操作方式,例如,在亲缘树上搜索自己感兴趣的物种与分类单元,系统会根据用户要求自动调整树的显示方式,以突出用户感兴趣的内容;结合其它生物学知识,用户可以对菌株的谱系属性试行调整,系统会根据新提交的谱系信息重新对比与标记亲缘关系树。CVTree3服务器还可以输出高质量的图以供展示和发表。有关交互操作的详细描述与操作方法可参阅在线手册。

|
| 树中带有实心圆点的枝表示可以通过点击进一步打开;空心圆圈则表示已经打开,点击则收缩;没有点/ 圈的枝则表示已经展开至最末端,不能进一步操作了;{} 内的数字表示基因组的数目的信息;红色表示CVTree 结果与参照分类信息符合的单源枝,蓝色表示它不是单源枝 图 3 由CVTree 方法得到的亲缘树并根据分类信息展开到门一级 |
物种亲缘关系与分类具有天然的联系,因此CVTree的一个重要应用:可以方便地使用亲缘关系对物种进行初步鉴定。在CVTree3服务器中,我们内置了大量已知分类信息的全基因组。用户只需要上传未知原核生物的全基因组数据,将它们和我们内置的全基因组数据混合生成亲缘树,就可以通过内置全基因组的谱系信息来推测上传的未知菌株的分类地位。
图 4所示也是CVTree3中Example的亲缘树。该树所使用的数据集与图 3相同,通过CVTree3的交互作用,我们让它更有效的显示“未知”物种的基因组的分类地位。我们从互联网下载了两个并没有包含在内建数据库之中的全基因组作为“未知”物种来进行测试。图 4中棕色显示的Kutzneria_albida_DSM_43870.UPLOAD{1}就是其中之一。该“未知”菌株,与Pseudonocardiaceae科的其它几个属的菌株同处于一个枝内,同时又与它们保持属一级的独立性,即与其它几个属相互并列。所以我们可以判定,上传的这个“未知”菌株应该是Pseudonocardiaceae科下的一个在CVTree3内建数据库中没有反映的“新”属。显然,这与我们从其它渠道了解到的信息,包括它的命名,是一致的。

|
| 标记为棕色的是上传的“未知”基因组,星号标记了“未知”基因组与某些内部基因组共同形成的枝所对应的一个分类单元 图 4 上传基因组与内部基因组的亲缘关系 |
基于未知菌株的全基因组序列,使用CVTree3可以对菌株进行亲缘与分类鉴别研究。虽然使用16S rRNA序列的联配也可以进行类似的研究,但是由于信息量的限制,使用16S rRNA方很难进行种以下的分类单元的研究,这正是CVTree方法的优势所在。由于CVTree方法合理的利用了全基因组信息,它的分辨率显著高于16S rRNA,从而可以进行种以下亲缘关系的研究。下面我们以冰岛硫化叶菌(Sulfolobus islandicus)来说明这种应用。
硫化叶菌是一类极端嗜热嗜酸古菌,多存在于地热泉、火山热泉与泥浆喷口处。冰岛硫化叶菌因最早发现于冰岛而得名。由于环境限制,在演化上相对隔绝,所以不同采集地的菌株基因组,具有一些不同的特征。在CVTree3的内建数据库中,共收集了10个属于该物种的菌株,它们来自4个不同的采集地。如图 5所示,在由CVTree方法建立的亲缘关系也显著地表现出其采集地的地理位置。首先,来自美洲的4个菌株与来自欧洲的6个菌株分成两个大枝,然后再根据其采集地的不同,进一步分成为4个较小的分枝。也就是说,从CVTree得到的亲缘关系,有效地反应了地理隔绝带来的演化效果[24]。除了这种由地理位置带来的种以下的分化外,种以下的分类单元,如亚种、生态型、血清型、生物变种等,在CVTree构建的亲缘关系树中得以体现的例子,在我们的研究过程中还遇到很多。但是需要说明的是,这些因素之间又会交互影响。例如,相同亚种的不同血清型与相同血清型的不同亚种,对于化脓性链球菌在CVTree上有较好的关联,而对于肺炎链球菌就不那么清晰,还需要结合专业知识有针对性地进行研究。

|
| 颜色标记了菌株的采集地,泛红色代表美洲,泛蓝色代表欧洲 图 5 十个冰岛硫化叶菌(Sulfolobus islandicus)菌株与其采集地 |
在以上两个例子中,我们使用CVTree方法作为独立的检测工具,重现了与其它研究一致的结果。实际上基于CVTree方法,我们还能对现有的分类系统提供某些新的具有建设性的意见。例如,大肠杆菌(Escherichia coli)与志贺氏痢疾杆菌(Shigella),这两类肠道菌在形态上非常相似,都是革兰氏阴性杆菌,但是由于志贺氏痢疾杆菌在病理学上的特异性,它们被单独分类为一个属。另一方面,在基于部分基因的分类研究中,各种志贺氏痢疾杆菌常常与埃希氏细菌属下的大肠杆菌混杂在一起[25, 26],很多人就据此认为志贺氏痢疾杆菌与大肠杆菌应该是同一个种下的不同菌株。
我们用CVTree研究了埃希氏菌属与志贺氏痢疾杆菌属。如图 6所示,所有的志贺氏痢疾杆菌属的菌株都插入了埃希氏杆菌的属中,它们与大肠杆菌最靠近,但是也同所有的大肠杆菌分开。所有的志贺氏痢疾杆菌也单独分开。它们之间的分界也是明确的。这就表明志贺氏痢疾杆菌与大肠杆菌并非同种,它们属于埃希氏菌属,是大肠杆菌的姊妹种[27]。这是与目前流行观念不一致的看法,但它既区分了志贺氏痢疾杆菌与大肠杆菌,又反映了前者在病理上的特殊性。这说明CVTree的高分辨力对于医学实践中致病菌的检测会有所帮助。

|
| 亲缘关系展开至种的级别,绿色标记了志贺氏痢疾杆菌属,红色标记埃希 氏菌属 图 6 志贺氏痢疾杆菌属(Shigella)与埃希氏菌属 (Escherichia)的亲缘关系树 |
当前测序技术的革新带来了海量的基因组数据,为基于全因组数据的数据分析提供了丰富素材,同时也对发展合适的计算工具带来挑战。各种计算瓶颈与人为选择所导致的差异促使我们去研究无参数和不依靠序列联配的方法。基于全基因组的CVTree方法的提出与改进,就是在这一前提下的努力结果。它合理地利用了全基因组的信息,高效地实现了基于全基因组的亲缘关系和分类系统研究。它一方面能与传统的分类系统保持较好的一致性,另一方面它还提供了研究种以菌株的分辨能力,并且为解决一些具有特殊分类需求的问题提供帮助。我们新开发的CVTree3网络服务器,运行在并行的高性能硬件上,是一款高效与方便的基于全基因组的亲缘关系与分类系统的研究工具。它的使用界面非常友好,实现了亲缘关系与分类系统的自动比较,允许用户在浏览器上进行交互式操作。随着测序技术的提高,菌株测序的成本不久将低于鉴定它的“湿”实验的预算,生物工作者今后不必进行太多的鉴定实验,只要拿到菌株的全基因组,将它提交到CVTree3网络服务器上,就可以对它的分类特性进行初步判定。我们更希望,CVTree方法将来能够成为阐明原核生物亲缘关系与分类系统的定义性的工具。
致谢:感谢戚继、徐昭博士对CVTree网络服务器2004和2009版本的贡献以及参与CVTree3的讨论。感谢复旦大学物理系和应用表面物理国家重点实验室资助购进用于CVTree3的并行集群系统,使得整个研究项目得以持续进行。
| [1] | Whitman WB, Coleman DC, Wiebe WJ . Prokaryotes:the unseen majority[J]. Proc Natl Acad Sci USA, 1998, 95:6578-6583. |
| [2] | Woese CR, Stackebrandt E, Macke TJ, Fox GE. A phylogenetic definition of the major eubacterial taxa[J]. Syst Appl Microbiol, 1985, 6:143-151. |
| [3] | Staley JT. The bacterial species dilemma and the genomic-phylogenetic species concept[J]. Philos Trans R Soc Lond B Biol Sci, 2006, 361:1899-1909. |
| [4] | Yarza P, Richter M, Peplies J, et al. The all-species living tree project:a 16S rRNA-based phylogenetic tree of all sequenced type strains[J]Syst Appl Microbiol, 2008, 31(4), 241-250. |
| [5] | Whitman WB. Intent of the nomenclatural code and recommendations about naming new species based on genomic sequences[J]. Bull Bergey’s Int Soc Microb Syst, 2011, 2:135-139. |
| [6] | Wu D, Hugenholtz P, Mavromatis K, et al. A Phylogeny-driven genomic encyclopaedia of Bacteria and Archaea[J]. Nature, 2009, 462:1056-1060. |
| [7] | Goffeau A. Life with 482-Genes[J]. Science, 1995, 270:445-446. |
| [8] | Schneiker S, Perlova O, Kaiser O, et al. Complete genome sequence of the myxobacterium Sorangium cellulosum[J]. Nat Biotechnol, 2007, 25:1281-1289. |
| [9] | Qi J, Wang B, Hao B. Whole proteome prokaryote phylogeny without sequence alignment:a k-string composition approach[J]. J Mol Evol, 2004, 58:1-11. |
| [10] | Gao L, Qi J, Wei H, et al. Molecular phylogeny of coronaviruses including human molecular phylogeny of coronaviruses including human[J]. Chinese Sci Bull, 2003, 48:1170-1174. |
| [11] | Hao BL. A few pieces of mathematics inspired by real biological data.[M]//Ge ML, Oh CH, Phua KK. Proceedings of the Conference in Honor of C N Yang's 85th Birthday. World Scientific Pub Co Inc, 2008. |
| [12] | Hao BL, Gao L. Prokaryotic branch of the tree of life:a composition vector approach[J]. J Syst Evol, 2008, 46:258-262. |
| [13] | Hao BL, Long MY, Gu HY, et al. Whole-genome based prokaryotic branches in the tree of life[C]. Darwin 200 Beijing Int Conf, 2010:102-103. |
| [14] | Li QA, Xu Z, Hao B. Composition vector approach to whole-genome-based prokaryotic phylogeny:success and foundations[J]J Biotechnol, 2010, 149:115-119. |
| [15] | Zuo G, Xu Z, Hao B. Phylogeny and taxonomy of archaea:a comparison of the whole-genome-based CVTree approach with 16S rRNA sequence analysis[J]. Life, 2015, 5:949-968. |
| [16] | Wang H, Xu Z, Gao L, Hao B. A fungal phylogeny based on 82 complete genomes using the composition vector method[J]Bmc Evol Biol, 2009, 9:1471-2148. |
| [17] | Chu KH, Qi J, Yu ZG, Anh V. Origin and phylogeny of chloroplasts revealed by a simple correlation analysis of complete genomes[J]. Mol Biol Evol, 2004, 21:200-206. |
| [18] | Liu J, Wang H, Yang H, et al. Composition-based classification of short metagenomic sequences elucidates the landscapes of taxonomic and functional enrichment of microorganisms[J]. Nucleic Acids Res, 2013, 41:1-10. |
| [19] | Qi J, Luo H, Hao B. CVTree:A phylogenetic tree reconstruction tool based on whole genomes[J]Nucleic Acids Res, 2004, 32:45-47. |
| [20] | Xu Z, Hao BL. CVTree Update:A newly designed phylogenetic study platform using composition vectors and whole genomes[J]Nucleic Acids Res, 2009, 37:W174-W178. |
| [21] | Zuo G, Hao B. CVTree3 web server for whole genome-based and alignment-free prokaryotic phylogeny and taxonomy[J]Genomics Proteomics Bioinforma, 2015, (in press). |
| [22] | Saitou N, Nei M. The neighbour joining method:a new method for reconstructing phylogenetic trees[J]Mol Biol Evol, 1987, 4(4):406-425. |
| [23] | MihaescuR, Levy D, Pachter L. Why neighbor-joining works[J]. Algorithmica(New York), 2009, 54:1-24. |
| [24] | Zuo G, Hao B, Staley JT. Geographic divergence of ‘sulfolobus islandicus’ strains assessed by genomic analyses including electronic DNA hybridization confirms they are geovars[J]. Antonie Van Leeuwenhoek, 2014, 105(2):431-435. |
| [25] | Brenner DJ, Fanning GR, Miklos GV, Steigerwalt AG. Polynucleo-tide sequence relatedness among Shigella species[J]. Int J Syst Bacteriol, 1973, 23:1-7. |
| [26] | Brenner DJ, Fanning GR, Skerman FJ, Falkow S. Polynucleotide sequence divergence among strains of Escherichia coli and closely related organisms[J]. J Bacteriol, 1972, 109:953-965, 1972. |
| [27] | Zuo G, Xu Z, Hao B. Shigella strains are not clones of Escherichia coli but sister species in the genus Escherichia[J]Genomics Proteomics Bioinforma, 2013, 11:61-65. |