




2. 江南大学生物工程学院 工业生物技术教育部重点实验室江南大学,无锡 214122
2. School of Biotechnology and Key Laboratory of Industrial Biotechnology Ministry of Education,Jiangnan University,Wuxi 214122
N-乙酰转移酶(Mpr1)是能够乙酰化脯氨酸类似物2-羧酸氮杂环丁烷(AZC)的一种胞内酶[1]。酿酒酵母(Saccharomyces cerevisiae)∑1278b中的Mpr1可以使AZC乙酰化成N-acetyl AZC,从而防止AZC被误认为脯氨酸被生物体利用而产生毒性[2]。近年的研究发现Mpr1酶具有显著的抗活性氧簇(ROS)氧化胁迫生理功能[2, 6]。ROS是一类包含O2·、H2O2和·OH等的小分子物质,是正常细胞的新陈代谢的副产物。ROS化学性质极为活泼,积累后会给细胞造成极大的危害,导致细胞蛋白质发生解聚与降解[3]; 还能同细胞器膜中不饱和脂肪酸发生反应,导致细胞器功能受损[4]; 严重时还能造成DNA的断裂,引起细胞死亡[5]。在合成精氨酸途径中Mpr1可直接将Δ1-吡咯啉-5-羧酸酯(P5C)或其平衡物谷氨酸-γ-半醛(GSA)乙酰化成N-乙酰GSA,从而减少了通常途径中的多个转化步骤,有利于精氨酸快速合成[6],提高细胞抗ROS的性能。目前研究都集中在来源于S. cerevisiae∑1278b的Mpr1基因上[2]。
毕赤酵母(Pichia pastoris)是一种应用极为广泛的真核表达系统,已表达了超过500多种异源蛋白质[7]。由于P. pastoris代谢甲醇的生理特性,在利用甲醇诱导发酵过程中产生大量的ROS而受到异常强烈的氧化胁迫。P. pastoris中一种N-乙酰转移酶(Mpr1)具有抗氧化胁迫功能,相关研究尚为空白。研究P. pastoris中的Mpr1抗氧化胁迫机制将有利于进一步优化P. pastoris细胞表达功能。本研究将来源于P. pastoris GS115的Mpr1基因在E. coli JM109进行重组表达,并利用响应面分析法对发酵过程中的诱导培养温度、IPTG诱导浓度、起始诱导OD进行优化,获得最佳产酶条件; 另外,对酶学性质进行初步研究; 同时分析Mpr1对原核细胞的生长影响。
1 材料与方法 1.1 材料 1.1.1 菌株P. pastoris GS115、E. coli JM109均由本实验室保藏,PQE30质粒购自北京鼎国昌盛生物技术有限责任公司。
1.1.2 培养基LB培养基(g/L):分子级蛋白胨5,酵母粉5,NaCl 10。
TB培养基(g/L):蛋白胨12,酵母粉24,甘油5,K2HPO4·3H2O 16.43,KH2PO4 2.34。
1.1.3 酶及其它试剂限制性内切酶BamH I、Sac I,T4 DNA连接酶、Taq酶及其配套产品、胶回收试剂盒,质粒提取试剂盒,E. coli感受态细胞制备试剂盒,酵母基因组提取试剂盒购于大连宝生物公司。DTNB、酵母无氨基基础氮源培养基购自上海生工。AZC、乙酰辅酶A购自Sigma。蛋白标准分子量以及SDS-PAGE试剂盒购于碧云天生物技术研究所。其它常规试剂购自国药集团。
1.2 方法 1.2.1 Mpr1基因的PCR扩增P. pastoris GS115基因组按照上海生工基因组提取试剂盒操作步骤提取。根据NCBI网站Mpr1基因序列(登录号:GENban XP_002492064.1)设计引物如下:P1(正向引物)5'-GACGGATCCATGAGTTCTACTCTAGATCCTGAAC-3'; P2(反向引物)5'-AACGAGCTCTTAAGAAAGGTCCGTATCGCATGT-3',下划线分别表示BamH I和Sac I酶切位点,以引物P1、P2对P. pastoris GS115基因组PCR扩增获得Mpr1基因片段。PCR反应体系50 μL:G+C buffer 10 μL,ddH2O 33.5 μL,PrimeSTAR® HS DNA Polymerase 0.5 μL,基因组1 μL,正向引物0.5 μL,反向引物0.5 μL,dNTP 4 μL。PCR反应条件:94℃预变性4 min; 98℃变性10 s,55℃退火5 s,72℃延伸90 s,30个循环; 72℃延伸10 min,4℃保温。1%琼脂糖凝胶电泳检测目的产物。
1.2.2 重组表达载体的构建使用胶回收试剂盒回收纯化PCR产物,于16℃与pMD18-T载体连接过夜后,转入E.coli JM109的感受态细胞中,挑取阳性单克隆菌落于液体LB培养基37℃培养8 h左右,抽提质粒,酶切验证后并进行基因测序。测序正确后将pMD18-T-Mpr1经BamH I、Sac I双酶切之后,将片段纯化、回收,质粒PQE30也以同样的方法酶切并胶回收。T4 ligase连接目的基因和载体,过夜后转入E.coli JM109的感受态细胞中,涂布于含有100 μg/mL Amp抗性浓度的LB平板,挑取单菌落于LB液体培养基中,37℃培养8 h左右,抽提质粒,酶切验证并测序,得到正确的表达载体命名为PQE30-Mpr1,获得重组菌命名为PQE30-Mpr1-E.coli JM109。
1.2.3 Mpr1酶基因的诱导表达将构建好的重组菌PQE30-Mpr1-E.coli JM109接种到含有Amp的LB培养基中,培养8 h后转接到含Amp的TB培养基,在37℃ 200 r/min生长至设定OD后加入IPTG在不同的温度下诱导表达。发酵后12 000 r/min离心5 min收集菌体,用Tris-HCl缓冲液(50 mmol/L,pH7.5)悬浮菌体,超生破碎菌体,12 000 r/min离心5 min后上清即为重组Mpr1酶的粗酶液。
1.2.4 N-乙酰转移酶Mpr1酶活测定[8] 在30℃条件下将1 mL反应液在412 nm下测定光吸收的变化。其中TNB的消光系数为15 570 M-1·cm-1。一个酶活力单位定义为在1 min内形成1 µmol TNB的量,具体成分包括:Tris-HCl buffer(pH7.5)50 mmol/L,800 μL; AZC 5 mmol/L,50 μL; acetyl-CoA 0.2 mmol/L,50 μL; DTNB 1 mmol/L,50 μL; 酶液50 μL。
反应计算公式:A = ε b c
其中,ε=15 570 M-1·cm-1,A为吸光度,b为测定物质浓度M(mol/L),c为液层厚度(cm)。
通过测定1 min内A的变化算出b的浓度的生成量,计算出酶活。
1.2.5 E. coli细胞内ROS含量测定用配制的磷酸钠缓冲溶液将离心的发酵液样品稀释为菌液OD600约0.2(细胞浓度1×107 cells/mL)的悬浮液,取1 mL菌液加入5 μL DCFH-DA溶液,在37℃、50 r/min水浴摇床内避光负载30 min,然后将样品放在冰上避光保藏[9]。对照样品加入5 μL不含DCFH-DA的DMSO,相同条件下处理; 用流式细胞技术(Flow cytometry,FCM)检测细胞内活性氧含量。
1.2.6 响应面优化实验Box及其合作者于20世纪50年代提出的响应面分析法(Response Surface Analysis)被广泛应用于食品、化工、农业等多个领域[10, 11]。本研究根据正交实验结果选取3个主要因素进行响应面实验设计,并用SAS 8.1软件对实验数据进行回归分析,通过微分方法预测实验最佳点。
1.2.7 重组Mpr1酶最适反应pH和pH稳定性分析 1.2.7.1 Mpr1酶最适反应pH将纯化获得酶液离心处理,用不同pH值的缓冲液复溶,在30℃下测定重组Mpr1酶活力,将最高的Mpr1酶活为定义为100%,并计算其它样品的相对活力。
1.2.7.2 Mpr1酶的pH稳定性将离心处理的酶用pH分别为3.0、4.0、5.0、6.0、6.5、7.0、7.5、8.0、8.5和9.0的50 mmol/L缓冲液复溶,于4℃放置24 h,测定其酶活力。
1.2.8 重组Mpr1酶最适反应温度和温度稳定性分析 1.2.8.1 Mpr1酶的最适反应温度将酶反应体系分别在15℃、20℃、25℃、30℃、35℃、40℃、45℃、50℃和60℃等温度下测定酶活力,将最高酶活定义为100%,并计算各样品的相对活力。
1.2.8.2 Mpr1酶的热稳定性将酶液分别在4℃、10℃、20℃、30℃和40℃等不同梯度温度条件下水浴保温,每隔1 h取一次样,测定残留酶活。
2 结果 2.1 重组表达载体的的构建提取P. pastoris GS115基因组DNA,PCR扩增得到基因Mpr1(图 1),将得到cDNA克隆到pMD18-T simple vector。测序结果表明,MPR基因全长627 bp,编码208个氨基酸,与NCBI网站报道的登录号为GENban XP_002492064.1的P. pastoris GS115的Mpr1编码的蛋白序列完全一致。

|
| M:DL2000 DNA Marker; 1,2:Mpr1基因 图 1 图1 PCR产物电泳图 |
将pMD18-T simple vector和表达载体PQE30分别进行BamH I、Sac I双酶切之后,胶回收、连接、转化E.coli JM109,挑取单克隆培养过夜后,抽提质粒,经双酶切得到大小约为3 460 bp和627 bp的DNA片段(图 2),获得的重组质粒PQE30-Mpr1,获得重组菌命名为PQE30-Mpr1-E.coli JM109。

|
| M1:λ-EcoT14 I digest DNA Marker; M2:DL2000 DNA Marker; 1,2:PQE30-Mpr1经BamH I和Sac I 双酶切 图 2 重组质粒PQE30-Mpr1酶切电泳验证 |
将重组菌PQE30-Mpr1-E.coli JM109接种到含Amp的LB培养基中,转接TB培养基诱导24 h后破壁胞内上清酶活为462 mU/mL,携带空载体的对照菌PQE30-E.coli JM109未检测到酶活。SDS-PAGE(图 3)显示,23 kD处有可溶性目的蛋白条带。

|
| M:蛋白Marker; 1-3:intracellular proteins of PQE30-E.coli JM109; 4-6:intracellular proteins of PQE30-Mpr1-E.coli JM109 图 3 重组菌发酵产Mpr1酶SDS-PAGE图 |
根据Box-Bchnken的中心组合设计原理[10],设计了三因素三水平的响应面分析实验,共有15个试验点,以发酵温度,IPTG含量和诱导起始OD为自变量,以发酵液酶活为响应值,实验因素与水平的选取,见表 1。
15个实验点可分为两类:一是析因点,自变量取值在x1,x2,x3所构成的三维顶点,共12个析因点; 二是零点,为区域的中心点,零点实验重复3次,以估计实验误差,每次实验得到的酶活,见表 2。
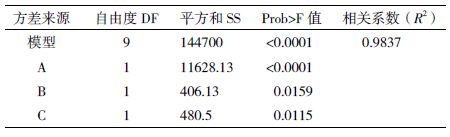
以发酵液酶活产量为响应值,运用SAS程序进行回归拟合后,各实验因子对响应值的影响可用下列函数表示:Y1=604.67-38.13X1+7.12X2+7.75X3-2.75X1X2-0.5X1X3+1X2X3-187.96X12-3.46X22-32.21X32。运用SAS程序对回归方程进行方差分析,结果(表 3)显示,所做模型和3个因素的“Prob>F”值远小于0.05,说明所做的回归方程模型及3个影响因素的结果是显著的。根据R2=0.983 7能解释98.37%的发酵酶活变化水平。因此回归方程为重组E. coli发酵产Mpr1酶提供了一个适合的模型。计算得到的RSA(图 4)显示,在3种因素中对发酵酶活影响由大到小分别是诱导温度>IPTG浓度>起始诱导生物量,说明温度是影响重组E. coli诱导产酶最关键因素。经RSA拟合后可看到拟合模型具有真实的最大值。

|
| 图 4 重组Mpr1酶活与诱导温度(A)、起始诱导生物量(B)和IPTG浓度(C)的关系 |
经过模型计算预测得到最大发酵酶活产量为611.7,其中3个因素的最佳值分别是诱导温度在30℃,起始诱导生物量OD为4.5,IPTG浓度控制在0.4 mmol/mL。为了证实预测的结果,用以上得到的最优配方重复实验3次,平均发酵酶活为(610.3±9.5)mU/mL,与预测值611.7良好的拟合性证实了模型的有效性,回归方程为摇瓶发酵产Mpr1酶提供了一个合适的模型。
2.3 重组Mpr1酶酶学性质初步研究将细胞高压破碎后离心收集上清液,通过硫酸铵沉淀、透析、DEAE阴离子交换层析获得纯化的Mpr1酶,进行了酶学性质初步研究。
2.3.1 重组Mpr1酶的最适反应pH和pH稳定性重组Mpr1酶反应的最适pH的研究结果(图 5-A)显示,pH在7-7.5酶活性最高。考察重组Mpr1酶的pH稳定性,将其置于pH3.0-9.0的缓冲体系中4℃保温24 h后,测定残留酶活。结果(图 5-B)显示,重组Mpr1酶的pH稳定范围为6.5-8.0,在此范围内保温24 h,重组Mpr1酶活约保持在90%以上,在pH4.0-6.5范围内保温24 h,酶活可保持在50%以上,而当pH>8.0和pH<4.0时,Mpr1酶稳定性很差。

|
| 图 5 Mpr1 酶反应的最适pH(A)和pH 稳定性(B) |
在不同温度条件下进行酶促反应,测定Mpr1酶酶活力。结果(图 6-A)显示,重组Mpr1酶在30℃时酶活最高,而当温度继续上升后,Mpr1酶活力逐渐下降,可能是由于高温造成了酶蛋白逐渐变性。将重组Mpr1酶分别在不同温度条件下水浴保温,考察其热稳定性。结果(图 6-B)显示,重组Mpr1酶在4-20℃之间,保温3 h Mpr1酶的表现相对稳定,在30℃保温3 h,酶活残留50%左右,而在40℃下保温2 h剩余活性不足40%,表明重组Mpr1酶在低温条件下具有较好的稳定性。

|
| 图 6 Mpr1 酶的最适反应温度(A)和热稳定性(B) |
Momura等[12]在研究来源于S. cerevisiae细胞∑1278b和粟酒裂殖酵母(Schizosaccharomyces pom-be)的N-乙酰转移酶时,发现将上述来源的N-乙酰转移酶在重组E. coli中实现异源表达后,重组E. coli同时还具有了抗AZC的活性。在利用重组E. coli胞内表达P. pastoris来源Mpr1时发现重组菌的生长状况要好于对照菌(图 7),在发酵24 h后重组菌的细胞OD达到14左右,而对照菌OD仅为9。基于Mpr1具有显著的抗氧化胁迫的功能,又测定了E. coli胞内ROS水平,结果(表 4)发现重组菌胞内ROS含量显著低于对照菌。这可能是重组E. coli表达Mpr1时,Mpr1酶使其抗氧化胁迫能力得到增强,从而改善细胞生长环境,因而生长速度明显高于对照组。

|
| 图 7 PQE30-E.coli JM109菌株和PQE30-Mpr1-E.coli JM109菌株细胞生长情况比较 |
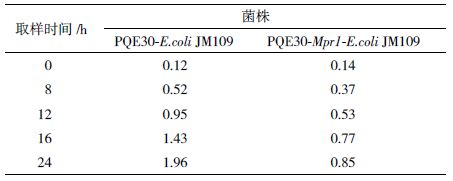
目前国外研究报道均集中于S. cerevisiae的Mpr1。P. pastoris应用广泛,但是其在诱导的过程中受到的氧化胁迫严重影响其外源蛋白的表达性能,有关P. pastoris MPR1基因的研究目前尚属空白。由于MPR1基因较为广泛的分布于生物细胞中[13],而所有的生物细胞都会受到ROS的影响,因此研究MPR1的表达调控也是抗氧化胁迫机制的共性问题之一。但是,毕赤酵母Mpr1在酵母内表达量很低,无法满足研究的需求,需要通过基因工程手段提高Mpr1酶的表达量。本实验在E.coli JM109中成功实现胞内表达来源于P. pastoris GS115的Mpr1酶。通过研究酶学性质,P. pastoris Mpr1和S. cerevisiae Mpr1相比较具有较大区别。Michiyo等[12]研究同样在E. coli中表达的S. cerevisiae细胞∑1278b中Mpr1酶时,发现其最适反应pH范围在8.5-9.0。可见来源于P. pastoris的Mpr1酶更适合于在偏中性的环境下发挥酶功能。Iinoya等[8]发现S. cerevisiae Mpr1酶在50℃条件下半衰期为8.2 min,相比之下本研究中P. pastoris Mpr1在40℃下半衰期为1 h左右,可见P. pastoris来源Mpr1较S. cerevisiae来源的Mpr1更加稳定。经NCBI网站blast比对分析发现,两种同工酶的同源性仅为47%。鉴于P. pastoris具有极强的抗氧化胁迫能力,其Mpr1酶可能在抗氧化胁功能上存在特殊性,或者还存在有不同的生理功能,需要进一步实验来明确P. pastoris中Mpr1的生理功能。
为了获得Mpr1酶的最佳发酵条件,采用RSA方法分别从诱导温度、IPTG诱导浓度、起始诱导OD进行发酵优化,最终酶活达到(610.3±9.5)mU/mL。优化发现发酵温度对发酵水平的影响最大,其次是IPTG浓度和起始诱导OD。由于在利用E. coli异源表达时温度越高,蛋白表达越容易形成包涵体,因而发酵温度是影响蛋白表达的重要因素。较低发酵温度有利于细胞控制自身的合成蛋白质的速率,从而表达更多具有正常生理功能的蛋白质。另外,本研究中还发现PQE30-Mpr1-E.coli JM109菌株的生长情况显著好于PQE30-E.coli JM109菌株,经分析各自胞内ROS 水平,认为是重组菌中所表达的Mpr1使其获得更高的抗氧化胁迫能力,从而具有更强的生长能力。鉴于P. pastoris的生理特殊性,后续研究可通过构建酵母Mpr1缺失突变体和过表达突变体,研究Mpr1在P. pastoris中的抗氧化胁迫调控规律,在分子水平阐明其抗ROS氧化胁迫的调控机制。
4 结论构建了重组菌PQE30-Mpr1-E.coli JM109,采用RSA方法从诱导温度、IPTG诱导浓度、起始诱导OD分别对其进行发酵优化,获得最佳诱导发酵条件为:30℃,起始诱导OD=4.5,0.4 mmol IPTG诱导24 h后,Mpr1酶活达到(610.3±9.5)mU/mL。酶学性质初步研究结果表明,Mpr1酶的最适反应pH范围为7.0-7.5,最适反应温度是30℃。此外,对比重组菌和对照菌在同样条件下培养,重组菌生物量显著高于对照菌,胞内ROS含量较低,说明将Mpr1酶在E.coli JM109中表达之后,提高了其抗ROS胁迫能力,促进了细胞的生长。
| [1] | Takagi H, Shichiri M, Takemura M, et al. Saccharomyces cerevisiae _1278b has novel genes of the N-acetyltransferase gene superfamily required for L-proline analogue resistance[J]. J Bacteriol, 2000, 182:4249-4256. |
| [2] | Shichiri M, Hoshikawa C, Nakamori S, et al. A novel acetyltransfer-ase found in Saccharomyces cerevisiae_1278b that detoxifies a proline analogue, azetidine-2-carboxylic acid[J]. J Biol Chem, 2001, 276:41998-42002. |
| [3] | Gebicki S, Gill KH, Dean RT, et al. Action of peroxidases on protein hydroperoxides[J]. Redox Rep, 2002, 7:235-242. |
| [4] | Poljak A, Dawes IW, Ingelse BA, et al. Oxidative damage to proteins in yeast cells exposed to adaptive levels of H2O2[J]. Redox Rep, 2003, 8:371-377. |
| [5] | Lee J, Romeo A, Kosman DJ. Transcriptional remodeling and G1 arrest in dioxygen stress in Saccharomyces cerevisiae[J]. J Biol Chem, 1996, 271:24885-24893. |
| [6] | Sasano Y, Takahashi S, Shima J, et al. Antioxidant N-acetyltransfe-rase Mpr1/2 of industrial baker’s yeast enhances fermentation ability after air-drying stress in bread dough[J]. International Journal of Food Microbiology, 2010, 138:181-185. |
| [7] | Cos O, Ramon R, Montesinos JL, et al. Operational strategies, monit-oring and control of heterologous protein production in the methylot-rophie yeast Pichia pastoris under different promoters:a review[J]. Microb Cell Fact, 2006, 5:17. |
| [8] | Iinoya K, Kotani T, Sasano Y, et al. Engineering of the yeast antioxi-dant enzyme Mpr1 for enhanced activity and stability[J]. Biotech-nol Bioeng, 2009, 103:341-352. |
| [9] | 肖安风, 周祥山, 周利, 张元兴. 流式细胞术检测毕赤酵母发酵过程中胞内活性氧水平[J]. 生物工程学报, 2006, 22(2):273-277. |
| [10] | Thompson DR. Response surface experimentation[J]. J Food Proc Pres, 1982(6):155. |
| [11] | 葛宇, 许时婴, 王璋. 响应面优化冰淇淋复配稳定剂配方研究[J]. 食品科学, 1995, 16(11):5-9. |
| [12] | Michiyo N, Shigeru N, Hiroshi T. Characterization of novel acetyltransferases found in budding and fission yeasts that detoxify a proline analogue, azetidine-2-carboxylic acid[J]. J Biochem, 2003, 133:67-74. |
| [13] | Masaru W, Kae O, Michihiko K, et al. Distribution of L-azetidine-2-carboxylate N-acetyltransferase in yeast[J]. Biosci Biotechnol Biochem, 2008, 72:582-586. |