




DNA 甲基化是DNA 甲基转移酶(DNA methylt-ransferase,DNMTs)将S-2腺苷蛋氨酸(S-2 aden-osine methionine,SAM)上活化的甲基基团转移到DNA链中,是哺乳动物中最常见和研究得最为深入的表观遗传修饰。而胞嘧啶C-5位点的甲基化(5-甲基-脱氧胞苷,5mdC)是最主要的DNA甲基化形式[1],主要发生在CpG核苷酸序列中。人类基因组中绝大多数散在分布的CpG位点为甲基化修饰,而CpG岛(定义为长度大约1 kb,G+C含量大于55%并以二核苷酸CpG形式出现)通常为非甲基化状态(尤其与启动子区相关的CpG岛区域),并与基因的表达密切相关[2]。DNA 甲基化异常主要包括整体基因组和基因启动子区甲基化水平的改变,基因组甲基化改变导致染色质稳定性降低,基因启动子区甲基化改变则会干扰基因转录,使基因正常表达受损,可引起细胞的异常增殖和癌变[3]。此外,DNA 甲基化还与机体衰老、转座子沉默、X 染色体失活及基因印记的形成有关[1, 2, 3]。
近年来,DNA甲基化成为了生命科学的研究热点,研究方法不断发展,而以往建立在常规分子生物学技术基础之上的研究方法存在某些缺陷,如克隆和PCR扩增缺乏甲基转移酶,5mdC不能保存下来,且此修饰存在于DNA双链的大沟中而杂交技术难以识别等。此外,基于亲和富集/剔除、甲基化敏感的核酸内切酶消化或亚硫酸氢盐修饰的DNA预处理方法,已建立了一系列分析技术,它们能与其他的研究方法联合检测5mdC标记的存在和位点,例如测序、杂交、质谱检测、凝胶电泳和PCR等,它们在灵敏度、分辨率、重复性、误差、通量、成本和定量的程度上存在差异[4, 5]。质谱(Mass spectrometry)以高敏感性、高通量、高准确率、高分辨率、快速且重复性好而成为该领域研究的关键技术,尤其在检测大样本的微量甲基化时,高灵敏度、高通量和定量DNA甲基化分析方法是必需的,质谱联合液相色谱(Liquid chromatogram)等技术在这方面显示了巨大潜力。本文将分别阐述利用质谱和色谱联合质谱技术研究DNA甲基化的方法。
1 启动子区的甲基化检测基质辅助激光解析电离飞行时间质谱(Matrix-assisted laser desorption/ionization time of flight mass spectrometry,MALDI-TOF-MS)在遗传学、表观遗传学和核酸研究领域应用广泛,如单核苷酸多态性基因分型、突变检测、基因拷贝数变异和剪接变异检测等[6]。结合碱基特异性酶切反应与MALDI-TOF-MS检测原理,Sequenom公司的MassARRAY检测系统,是用于单核苷酸多态性分型和DNA甲基化定量检测的技术体系,具备高通量的检测性能,实现多重CpG位点的分析检测,可量化 <5%的甲基化改变[7],能用于基因组任何区域(如启动子区)或候选基因的DNA甲基化定量分析[8]。
如图 1所示,基因组DNA 经亚硫酸氢盐处理后,未甲基化的胞嘧啶转化为尿嘧啶(C→U),而甲基化的胞嘧啶保持不变;设计5'-端含T7启动子的引物扩增DNA双链,扩增片段长度一般为200-600 bp,碱性磷酸酶混合液去除反应体系中游离的核苷酸;再加入T7 RNA聚合酶等进行体外转录和碱基特异性的酶切反应;Nanodispenser从384孔板裂解反应液中取微量溶液转入基质芯片上,质谱仪收集质谱图[9]。

扩增产物完全的碱基特异性裂解可产生适合MALDI-TOF-MS检测范围的短小核苷酸片段,该系统所配备的数据库含参考序列的信息,由特定的算法计算序列的分子量改变并搜索相应的参考序列,从而发现序列的变化。由于C→T变化致使反义链中G→A变化,含有甲基化CpG 位点的片段与不含甲基化CpG位点的片段每个CpG 位点之间分子量相差16 Da。计算获得核苷酸片段中每个CpG单位的相对甲基化率(单个CpG 单位可含有多个CpG位点),EpiTYPER软件对各CpG 位点或CpG 单位进行甲基化定量分析。EpiTYPER系统提供先进方便的DNA甲基化定量分析手段,提供数字和图像注释工具并将实验数据与检测的核苷酸序列相匹配[9, 10](图 2和图 3)。


MassARRAY系统突出特点是能以极高的精确度快速进行基因型识别,直接获得序列的甲基化信息。研究者们对该技术平台不断优化,例如,采用不同碱基特异性的酶切反应,得到不同组合的核苷酸片段,目的序列的覆盖率增加,95%以上的序列能够被检测出[11]。Thomson设计了统计分析软件分析来自EpiTYPER数据以检验亚硫酸氢盐处理的效率的高低[12]。Wong等[13]用干燥的血样提取高纯度的DNA并进行高效的亚硫酸氢盐修饰,降低了样本要求,使该技术应用范围扩大。
该方法也存在一定不足,如碎裂的核苷酸片段中,只有单个碱基对的改变能够被软件推测出,更加复杂的突变和多态性则难以检出[14]。CG位点是常见的突变位点,甲基化的胞嘧啶经过脱氨基变成胸腺嘧啶,此多态性不能被亚硫酸氢盐处理所辨别,会导致DNA甲基化检测的错误[15]。SNP的存在会改变核苷酸片段断裂的模式和分子量,而EpiTYPER不能分辨出,会导致结果不准确。当核苷酸片段的分子量超出软件分析(1.5-7 kD)范围时,此核苷酸片段上一系列CG位点的信息难以获得;多个CG位点同时出现在同一核苷酸片段上且需要同时分析时,导致分辨率下降。PCR和亚硫酸氢盐处理也是实验误差的重要来源,获得纯净的PCR产物对于检测和分析至关重要。复杂组织中细胞类型的异质性或同质细胞表观遗传性状的不一致使得分析变得复杂,结果可靠性也会下降[16]。
Agrawal等[17]利用MassARRAY技术平台对80例急性骨髓性白血病病人骨髓样本的肿瘤抑制基因C/EBP基因启动子区分析,发现该基因启动子发生高甲基化且基因表达水平下降,说明该病的发生可能与此基因启动子高甲基化有关。Raval[8]发现,因启动子高甲基化导致死亡相关蛋白激酶1(DAPK1)基因沉默几乎发生在所有的慢性B淋巴细胞白血病病例中。在乳腺癌危险性评价[18],神经管缺陷的危险因素研究[19],慢性B淋巴细胞白血病[8]、结直肠癌[20]和孕妇产前无创性诊断[21]等方面,MassARRAY技术平台都已有研究成果。
除MassARRAY技术平台外,根据质谱对核苷酸的分析依赖其电荷状态的特性,Tost等[22]开发了一种称作GOOD assay分析样品的准备方法。亚硫酸氢盐处理样品和甲基化PCR 扩增目的片段后,碱性磷酸酶消化未反应的底物。因为引物3'-末端碱基有磷硫桥,底物为α-S-dNTP,延伸产物为磷硫骨架,而用磷酸二酯酶只能消化正常的磷酸骨架,磷硫骨架却能抗消化。烷基化中和磷硫骨架链后用MALDI-TOF-MS分析。该方法只需少量样品,可实现多通路检测,但实验步骤较多,并利用酶消化会致检测样品纯度降低、杂质多,还需建立标准曲线,不利于推广应用。基于肽核酸(Peptide nucleic acid,PNA)与DNA结合的特异性高、不易被核酸酶降解、杂交后热稳定性好并能在低盐状态下稳定存在等优点,Schatz等[23]建立了一种PNA探针杂交结合MALDI-TOF检测研究CpG岛甲基化的技术,亚硫酸氢盐处理DNA样品后,扩增目的片段,并固定于固相介质上,与含有甲基化位点的PNA探针杂交,去除非特异性结合后,洗脱下来的探针直接进行MALDI-TOF-MS分析,分子量的大小与序列文库进行比对,获得序列的甲基化信息。该方法能多重平行分析多个甲基化位点,所需样品量小,但需设计探针,并建立标准曲线,实验步骤较多,限制了该技术的推广。
2 全基因组DNA甲基化检测研究基因组DNA甲基化水平的思路主要有:3H标记的甲基供体S-腺苷甲硫氨酸(3H-SAM)与DNA反应后,检测结合于DNA上的同位素标记的甲基基团[24],该方法需要的DNA样品量大,结果不能直接反映DNA双链中5mdC的比率;基于DNA甲基化限制性内切酶的5mdC含量检测[25],实验步骤繁琐且效率较低;应用特异性的抗5mdC抗体进行间接免疫荧光法检测,所需样品量大且精确性较低[26];酶解或酸解DNA双链为单个核苷,以色谱技术分离检测,或在色谱分离的基础上质谱检测。以色谱技术检测基因组DNA甲基化水平的方法主要有薄层色谱(TLC)[27]、气相色谱(GC)[28]、液相色谱(HP-LC)[29, 30]、毛细管电泳(CE)[31]等,此类方法可提供准确且重复性较好的结果,但也有不足。薄层色谱实验的花费较少,但准确度不高且需大量的DNA样品;气相色谱实验数据准确,但核苷是强极性的化合物,进样前需进行衍生化处理;液相色谱能弥补气相色谱方法的缺陷,但紫外检测器不能选择性地检测化合物,这要求将DNA水解后所有的核苷达到全部分离,或至少使目标化合物与其他核苷完全分离,且紫外检测器灵敏度有限,DNA样品量要求大;毛细管电泳的分离效率较高,但DNA水解样品中的基质对实验的重复性影响很大。
色谱串联质谱法分析基因组DNA甲基化的基本原理是用酶或酸分解DNA链为单个核苷,色谱分离的基础上采用质谱检测器测定核苷含量,样本需要量少,灵敏度高,质谱技术的发展使大规模、高通量、全自动检测分析成为可能。Rossella等[32]报道的基于色谱串联质谱技术分析全基因组DNA甲基化水平的方法主要有气相色谱串联质谱(GC-MS)和液相色谱串联质谱(HPLC-MS/MS),Zhang等[33]报道采用亲水相互作用色谱串联质谱(HILIC-MS),但此方法使用很少。由于DNA核苷为强极性化合物,因此采用GC-MS方法分析全基因组DNA甲基化时需要对核苷进行衍生化处理,衍生化步骤不仅繁琐,而且衍生化不充分会给结果带来极大误差。HPLC-MS/MS方法具有高灵敏性和选择性,且不需对核苷进行衍生化处理,给全基因组DNA甲基化定量分析带来了新的解决思路,实验步骤也相对简单(主要包括:优化色谱条件和质谱参数,用脱氧胞苷和5-甲基脱氧胞苷梯度标准溶液建立标准曲线,再将水解并超滤去蛋白的DNA样品上样检测,参照标准曲线获得甲基化率的大小)。现对HPLC-MS/MS检测全基因组DNA甲基化的方法概括,见表 1。
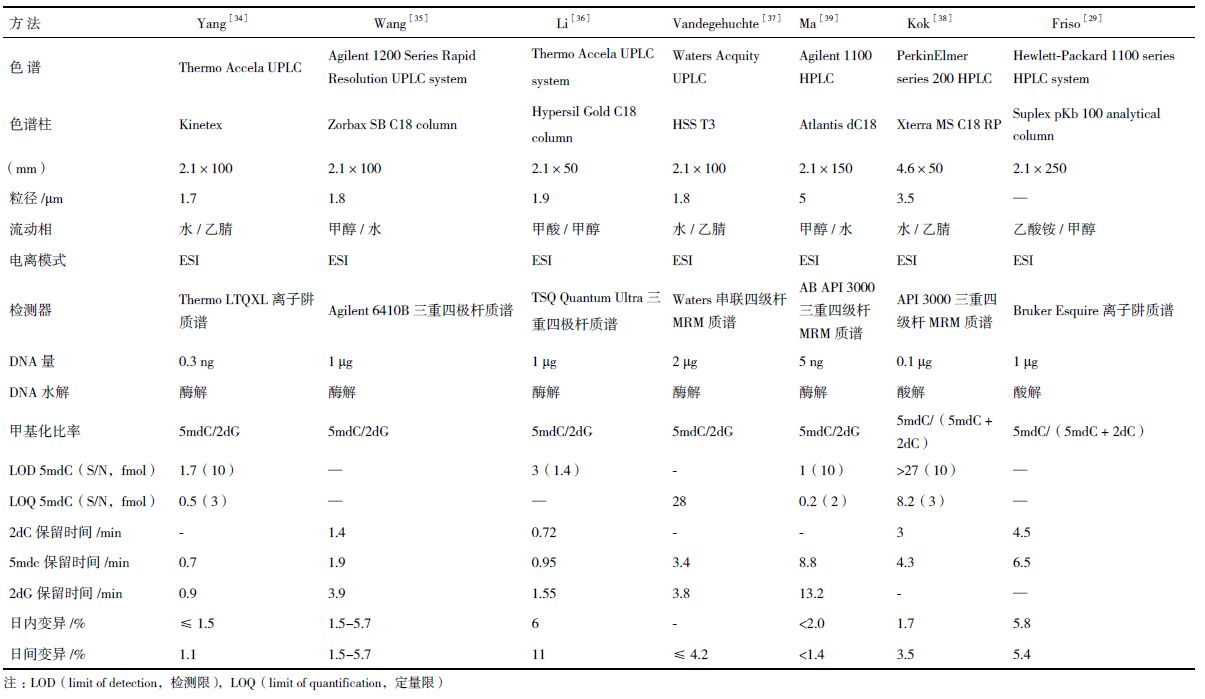
采用不同的色谱仪、色谱柱及质谱仪可建立不同的检测方法,各种方法各具特点。表 1中的方法强调提高敏感度(减少分析所需的DNA量,降低检出限)和通量,新方法的建立也以高通量、快速准确、价廉为目标。近年来,由于超高效液相色谱大幅度改善了液相色谱的分离度、样品通量和灵敏度,速度和分离度分别是传统HPLC的9倍和1.7倍,在低流速下,增加了峰浓度,提高离子源的效率,使灵敏度至少提高了3倍[33],并在液相色谱和质谱联用领域得到广泛发展。担负分离作用的色谱柱是色谱系统的关键,对色谱柱的要求是柱效高、选择性好、分析速度快,为提高分析的灵敏度并与质谱联用,发展了窄径柱、毛细管柱和内径 <0.2 mm的微径柱,能增加灵敏度,减少样品量,通过使用长柱达到高分离度,容易控制柱温。质谱检测器方面,Yang等[34]报道,三重四极杆质谱检测器比离子阱质谱检测器灵敏度更高,可避免连续分析中离子丧失的影响。
DNA甲基化定量也是该技术的关键之一,有文献报道直接采用外标法定量并建立标准曲线来度量甲基化率,另一些文献则采用昂贵的同位素内标15N3-dC和15N5-dG[36]、[15N3]2-dc和[methyl-d3,ring-6-d1]-5-methyl-2-dc[29],及生物合成的稳定同位素[U-15N]-dc和[U-15N]- 5mdC[43],它们分别代表 2dC和5mdC。Bagnyukova等[44]报道,HPLC-ICP-MS(电感耦合等离子体质谱)检测同位素31P和189Os(分别代表dC和5mdC)的量来定量甲基化比率。此外,有学者[34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45]采用5mdC和2-脱氧鸟苷(dG)物质的量比值法([5mdC]/[dG]比值法)进行定量,这种方法的原理是假设DNA双链中[5mdC]+[dC]=[dG],DNA分子中5mdC与dG物质的量的比值便可作为全基因组DNA甲基化计算公式的替代,不需考虑检测和分析中物质的损失,因为分子和分母比值分析会以相同的方式相互影响和抵消,可避免高成本同位素内部标准。但该方法在原理上也存在缺陷,即由于DNA分子中部分dG碱基会发生修饰,形成如O6-甲基化、C8-羟基化等修饰鸟苷,使得DNA分子中[5-mdC]+[dC]≈[dG],给准确性带来误差。总体来说,目前报道的HPLC-MS/MS技术分析全基因组DNA甲基化水平的方法几乎都未找到合适的内标来校正仪器误差并进行准确定量[36]。因此,还需进一步发展新型的基于液相色谱串联质谱检测全基因组DNA甲基化的方法。
现阶段,已有学者采用HPLC-MS/MS研究疾病与全基因组DNA甲基化的关联,例如,Lim等[43]用HPLC-ESI/MS研究无症状的大肠腺癌患者外周血白细胞全基因组DNA甲基化状况,发现全基因组DNA甲基化可能是大肠腺癌早期的致病因子。Bagnyukova等[44]研究发现,遗传毒性致肝癌物二乙基氨基芴致雄性SD大鼠肝脏组织全基因组DNA甲基化水平大幅度下降,但未在雌性小鼠的肝脏组织中发现此特征,而且雄性和雌性SD大鼠的肾脏中也不存在此特征。Choi等[45]用HPLC-ESI/MS检测Vitamin B-12缺乏致SD大鼠结肠DNA甲基化水平改变,初步认为这可能是肿瘤的诱因。
3 结语DNA甲基化作为重要的基因表达调控机制,不仅是当前生命科学和医学的研究热点,也在毒物的安全性评价中具有广阔的应用前景,其改变在许多毒物的毒理学机制中起着重要作用。近年来,已有学者研究毒物引起的DNA甲基化状态改变来探讨其可能的毒理学机制。例如,Tao等[46]研究发现,饮用水氯化消毒副产物(DBPs)引起的全基因组DNA和c-myc基因低甲基化的能力与它们致小鼠和大鼠肾脏和肝脏肿瘤及促进肿瘤发展的能力相关联。Pereira等[47]的研究表明,蛋氨酸作为甲基供体可抑制二氯乙酸(DCA)诱导的B6C3F1小鼠全基因组DNA甲基化水平下降,并抑制肝脏肿瘤进展,但未改变DCA所诱导的肝脏/体重比值和过氧化物酶增加,说明蛋氨酸对肝癌的抑制与其对DNA低甲基化的抑制有关,也说明了DNA低甲基化对于DCA诱导的肿瘤发生是至关重要的。DNA甲基化研究对毒物早期潜在毒效应的探测、实验浓度组的设置、剂量-反应曲线的有效定义和种属之间合适的浓度外推具有重大意义,将促进对毒物毒理学机制的研究,加快毒理学的发展[48]。在大规模基因组方面探索DNA甲基化特征,质谱以其高通量、高准确性和高灵敏度的特点将发挥着重要作用。熟悉其优势和不足是利用该技术平台的基础,随着自动化、微型化技术的发展和软件算法的改进,质谱技术将在高灵敏度、高精确性和低成本的基础上开展高通量的研究。
| [1] | Suzuki MM, Bird A. DNA methylation landscapes:provocative insights from epigenomics[J]. Nat Rev Genet, 2008, 9(6):465-476. |
| [2] | Tate P, Bird A. Effects of DNA methylation on DNA-binding proteins and gene expression[J]. Curr Opin Genet Dev, 1993, 3(2):226-231. |
| [3] | Clouaire T, Stancheva I. Methyl CpG binding proteins:specialized transcrip- tional repressors or structural components of chromatin[J]. Cell Mol Life Sci, 2008, 65(10):1509-1522. |
| [4] | Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences[J]. Nature, 2009, 462(727 1):315-322. |
| [5] | Ammerpohl O, Martín-Subero JI, Richter J, et al. Hunting for the 5th base:Techniques for analyzing DNA methylation[J]. Biochim Biophys Acta(BBA)-Gen Subj, 2009, 1790(9):847-862. |
| [6] | Ragoussis J, Elvidge GP, Kaur K, et al. Matrix-assisted laser desorption/ ionis ation, time-of-flight mass spectrometry in genomics research[J]. PLoS Genet, 2006, 2(7):e100. |
| [7] | CoolenMW, StathamAL, Clark SJ, et al. Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry:critical evaluation and improvements[J]. Nucleic Acids Res, 2007, 35:e119 |
| [8] | Raval A, Tanner SM, Byrd JC, et al. Downregulation of death-associated protein kinase 1(DAPK1)in chronic lymphocytic leukemia[J]. Cell, 2007, 129(5):879-890. |
| [9] | Ehrich M, Nelson MR, Stanssens P, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry[J]. Proc Natl Acad Sci, 2005, 102(44):15785-15790. |
| [10] | Watanabe T, Yachi K, Ohta T, et al. Aberrant hypermethylation of non-promoter zygote arrest 1(ZAR1)in human brain tumors[J]. Neurol Med Chir(Tokyo), 2010, 50(12):1062-1089. |
| [11] | Ehrich M, Hillenkamp F, Boom DVD. Perspectives in bioanalysis[M]. Elsevier, 2007. |
| [12] | Thompson RF, Suzuki M, Lau KW, et al. A pipeline for the quantitative analysis of CG dinucleotide methylation using mass spectrometry[J]. Bioinform Atics, 2009, 25(17):2164-2170. |
| [13] | Wong N, Morley R, Saffery R, et al. Archived Guthrie blood spots as a novel source for quantitative DNA methylation analysis[J]. Biotechniques, 2008, 45(4):423-424, 426, 428 . |
| [14] | Fryxell KJ, Moon WJ. CpG mutation values in the human genome are highly dependent on local GC content[J]. Mol Biol Evol, 2005, 22(3):650-658. |
| [15] | Ehrich M, Zoll S, Sur S, et al. A new method for accurate assessment of DNA quality after bisulfite treatment[J]. Nucleic Acids Res, 2007, 35(5):e29. |
| [16] | Mikeska T, Candiloro IL, Dobrovic A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation[J]. Epigenomics, 2010, 2(4):561-573. |
| [17] | Agrawal S, Hofmann WK, Tidow N, et al. The C/EBP delta tumor suppressor is silenced by hypermethylation in acute myeloid leukemia[J]. Blood, 2007, 109(9):3895-3905. |
| [18] | Radpour R, Kohler C, Haghighi MM, et al. Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array[J]. Oncogene, 2009, 28(33):2969-2678. |
| [19] | Wang L, Wang F, Guan J, et al. Relation between hypomethylation of long interspersed nucleotide elements and risk of neural tube defects[J]. Am J Clin Nutr, 2010, 91(5):1359-1367. |
| [20] | Kaneda A, Yagi K. Two groups of DNA methylation markers to classify colorectal cancer into three epigenotypes[J]. Cancer Sci, 2011, 102(1):18-24. |
| [21] | Bellido ML, Radpour R, Lapaire O, et al. MALDI-TOF mass array analysis of RASSF1A and SERPINB5 methylation patterns in human placenta and plasma[J]. Biol Reprod, 2010, 82(4):745-750. |
| [22] | Tost J, Schatz P, Schuster M, et al. Analysis and accurate quantification of CpG methylation by MALDI mass spectrometry[J]. Nucleic Acids Res, 2003, 31(9):e50. |
| [23] | Schatz P, Distler J, Berlin K, et al. Novel method for high throughput DNA methylation marker evaluation using PNA-probe library hybridization and MALDI-TOF detection[J]. Nucleic Acids Res, 2006, 34(8):e59. |
| [24] | Rampersaud GC, Kauwell GP, Hutson AD, et al. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women[J]. Am J Clin Nutr, 2000, 72(4):998-1003. |
| [25] | Yi P, Melnyk S, Pogribna M, et al. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocyst-eine and lymphocyte DNA hypomethylation[J]. J Biol Chem, 2000, 275(38):29318 -29323. |
| [26] | Santos F, Hendrich B, Reik W, et al. Dynamic reprogramming of DNA methyla tion in the early mouse embryo[J]. Dev Biol, 2002, 241(1):172-182. |
| [27] | Leonard GA, Hunter WN. Crystal and molecular structure of d(CGTAGATCTACG)at 2. 25 A resolution[J]. J Mol Biol, 1993, 234(1):198-208. |
| [28] | Gambari R, Barbieri R, Piva R, et al. Regulation of the expression of class II genes of the human major histocompatibility complex in tumor cells[J]. Ann N Y Acad Sci, 1987, 511:292-307. |
| [29] | Friso S, Choi SW, Dolnikowski GG, et al. A method to assess genomic DNA methylation using High-Performance Liquid Chromatogr- aphy/Electrospray Ionization Mass Spectrometry[J]. Anal Chem, 2002, 74(17):4526-4531. |
| [30] | 白桦, 邓大君. 引物延伸-变性高效液相色谱法检测去甲基化活化16型乳头状瘤病毒[J]. 中华预防医学杂志, 2007, 41(z1):81-83. |
| [31] | Wirtz M, Stach D, Kliem HC, et al. Determination of the DNA methylation level in tumor cells by capillary electrophoresis and laser-induced fluorescence detection[J]. Electrophoresis, 2004, 25(6):839-845. |
| [32] | Rossella F, Polledri E, Bollati V, et al. Development and validation of a gas chromatography/mass spectrometry method for the assessment of genomic DNA methylation[J]. Rapid Commun Mass Spectrom, 2009, 23(17):2637-2646. |
| [33] | Zhang JJ, Zhang LJ, Zhou KY, et al. Analysis of global DNA methylation by hydrophilic interaction ultra high-pressure liquid chromatography tandem mass spectrometry[J]. Analytical Biochemistry, 2011, 413(2):164-170. |
| [34] | Yang I, Fortin MC, Richardson JR, et al. Fused-core silica column ultra-performance liquid chromatographyion trap tandem mass spectrometry for determination of global DNA methylation status[J]. Nal Biochem, 2011, 409(1):138-143. |
| [35] | Wang X, Suo Y, Yin R, et al. Ultra-performance liquid chromatography /tand -em mass spectrometry for accurate quantification of global DNA methylation in human sperms[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2011, 879(19):1647-1652. |
| [36] | Li XN, Franke AA. High-throughput and cost-effective global DNA methylation assay by liquid chromatography-mass spectrometry[J]. Anal Chim Acta, 2011, 703(1):58-63. |
| [37] | Vandegehuchte MB, Lemiere F, Janssen CR. Quantitative DNA- methylation in Daphnia magna and effects of multigeneration Zn exposure[J]. Comp Biochem Physiol C Toxicol Pharmacol, 2009, 150(3):343-348. |
| [38] | Kok RM, Smith DE, Barto R, et al. Global DNA methylation measured by liquid chromatography tandem mass spectrometry:analytical technique, reference values, and determinants in healthy subjects[J]. Clin Chem Lab Med, 2007, 45(7):903-911. |
| [39] | Ma H, Zhang W, Hu J, et al. Analysis of global DNA methylation levels in human blood using high-performance liquid chromatography/tandem electrospray ionization mass spectrometry[J]. Eur J Mass Spectrom, 2009, 15(4):555-561. |
| [40] | Wang JB, Li HF, Jin C. Development and validation of a UPLC method for quality control of rhubarbbased medicine:Fast simultaneous determination of five anthraquinone derivatives[J]. Journal of Pharm aceutical and Biomedical Analysis, 2008, 47(4-5):765-770. |
| [41] | Quinlivan EP, Gregory III JF. DNA methylation determination by liquid chromatography-tandem mass spectrometry using novel biosynthetic[U-15N]deoxycytidine and[U-15N]methyldeoxycytidine internal standards[J]. Nucleic Acids Research, 2008, 36(18):e119. |
| [42] | Wrobel K, Landero Figueroa JA, Zaina S, et al. Phosphorus and osmium as elemental tags for the determination of global DNA methylation-a novel application of high performance liquid chromatography inductively coupled plasma mass spectrometry in epigenetic studies[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2010, 878(5-6):609-614. |
| [43] | Lim U, Flood A, Choi SW, et al. Genomic methylation of leukocyte DNA in relation to colorectal adenoma among asymptomatic women[J]. Gastroenterology, 2008, 134(1):47-55. |
| [44] | Bagnyukova TV, Tryndyak VP, Montgomery B, et al. Genetic and epigenetic changes in rat preneoplastic liver tissue induced by 2-acetylaminofluoren e[J]. Carcinogenesis, 2008, 29(3):638-646. |
| [45] | Choi SW, Friso S, Ghandour H, et al. Vitamin B-12 deficiency induces anomalies of base substitution and methylation in the DNA of rat colonic epithelium[J]. J Nutr, 2004, 134(4):750-755. |
| [46] | Tao LH, Wang W, Li L, et al. DNA hypomethylation induced by drinking water disinfection by-products in mouse and rat kidney[J]. Toxicol Sci, 2005, 87(2):344-352. |
| [47] | Pereira MA, Wang W, Kramer PM, et al. Prevention by methionine of dichloroacetic acid-induced liver cancer and DNA hypomethylation in mice[J]. Toxicol Sci, 2004, 77:243-248. |
| [48] | 杨建平, 朱志良, 袁建辉. DNA甲基化在毒理学中的应用前景[J]. 环境与职业医学杂志, 2007, 10(5):546-549. |