



自2000年起,本人在北京大学和中国农业科学院研究生院开设“实用生物信息技术”课程[1]。本课程以从事分子生物学实验研究的硕士或博士研究生为教学对象,重点介绍最基本、最常用的生物信息技术和方法,主要包括:(1)蛋白质和核酸序列相似性比对;(2)蛋白质序列数据库UniProt和核酸序列数据库RefSeq高级检索;(3)NCBI数据库相似性搜索工具Blast的应用;(4)利用MEGA软件构建分子系统发生树;(5)利用Swiss-PdbViewer软件显示、比较和分析蛋白质三维空间结构。
本文以人、小鼠、大鼠、斑头雁、灰雁几个不同物种的血红蛋白序列和结构为例,介绍这些常用生物信息技术和方法的具体应用。学生通过这些实例,能够初步掌握这些方法的具体应用,并能举一反三,将这些方法用于自己的课题研究,学会如何利用丰富的网络生物信息资源和分析工具解决自己正在进行或即将开始的研究课题中的实际问题。
1 序列比对 1.1 研究背景血红蛋白是人体血液中重要蛋白质分子,其主要生物学功能为运送氧气。血红蛋白分子为异源四聚体,可结合4个铁卟啉色素分子。成人血红蛋白分子由两个α-亚基和两个β-亚基组成。人类基因组中编码α-亚基的血红蛋白基因有两个,位于16号染色体短臂的α-珠蛋白基因簇中,其编码区核苷酸序列相同,所编码的蛋白质序列自然也相同,各含142个氨基酸残基。与人一样,小鼠和大鼠的血红蛋白也是四聚体,α-亚基也由142个氨基酸组成。小鼠和大鼠同属啮齿类动物,其共同祖先距今约2 500万年。而人属于灵长类动物,与啮齿类分歧时间约为9 500万年。对这3个物种α-血红蛋白氨基酸序列及其编码基因的核苷酸序列进行比对,可探索血红蛋白分子及其编码基因演化的特点。
1.2 比对方法和结果从国际蛋白质序列数据库UniProt中分别提取人和小鼠α-血红蛋白的FastA格式序列,其序列条目名称分别为HBA_HUMAN(人)、HBA_MOUSE(小鼠)。序列比对的软件很多,北京大学生物信息中心开发的综合序列分析平台WebLab(http://weblab.cbi.pku.edu.cn/)包括200多个程序[2]。利用WebLab中基于Needleman-Wunsch全局序列比对算法的程序Needle,采用默认蛋白质计分矩阵BLOSUM62和默认空位罚分值(起始空位罚分10.0,延伸空位罚分0.5),比对结果如图1所示。

|
| 图 1 人和小鼠血红蛋白α-亚基氨基酸序列比对输出结果 |
图 1中上方为统计值,包括序列长度(LENGTH)、比对分值(SCORE)、相同位点(IDENTITY)、相似位点(SIMILARITY)和空位数(GAPS)。下方为两条序列的具体比对结果,“|”表示相同位点、“:”表示相似位点,“.”表示不同位点。所谓相似位点,是指该位点的两个氨基酸理化性质较接近,如苏氨酸“T”和丝氨酸“S”、缬氨酸“V”和异亮氨酸“I”等。
按上述方法,分别对人/小鼠、人/大鼠、小鼠/大鼠3个物种α-血红蛋白进行序列比对,结果如表 1所示。
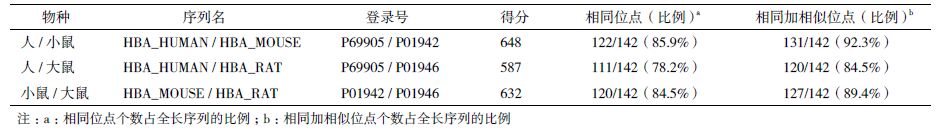
从NCBI参考序列数据库中提取这3个物种α-血红蛋白基因编码区序列,用WebLab中的Needle程序进行序列比对,注意选择核苷酸替换矩阵EDNAFULL,将起始空位罚分改为20.0,延伸空位罚分改为2.0,比对结果如表 2所示。
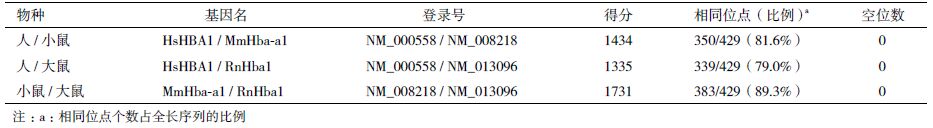
表 1为3个物种血红蛋白α-亚基氨基酸序列比对结果。出乎意料的是,人和小鼠α-血红蛋白共有122个相同位点,占全长142个位点的85.9%;而小鼠与大鼠之间的相同位点数为120个,占全长84.5%。换句话说,同为啮齿类的小鼠和大鼠,血红蛋白序列相似性低于啮齿类和灵长类。之所以出现这一结果,原因有许多,其中最主要的是密码子简并性,即同一氨基酸在不同物种或不同基因中可能由不同密码子编码,蛋白质序列相似性高低可能与其编码核苷酸的相似性高低并不一致。这3个物种血红蛋白编码基因的编码区核苷酸序列比对结果(表 2)显示,小鼠和大鼠之间的序列相似性为89.3%,高于小鼠和人之间的序列相似性81.6%。
2 数据库高级检索 2.1 研究背景研究表明,有些基因在一个物种中只有一个拷贝,称单拷贝基因,而真核生物基因组中大部分基因按基因家族形式存在,有多个拷贝,它们或者分布在同一染色体上相邻区域,或者分散在整个基因组不同染色体上。基因家族的产生包括全基因组水平重复和染色体片段重复等多种机制,是生物演化重要途径。同一家族的基因往往具有相似生物学功能,通过复杂的调控机制,在不同组织、不同环境或不同发育阶段表达。例如,无脊椎动物的血红蛋白由一个基因编码,而脊椎动物的血红蛋白则由多个基因编码。以人血红蛋白基因家族为例,分为α-珠蛋白(α-globin)和β-珠蛋白(β-globin)两个基因簇,如图 2所示。图 2上方为α-珠蛋白基因簇,位于16号染色体短臂正链150-180 kb区段,全长约30 kb,按5'-3'顺序依次为ζ、μ、α2、α1和θ-珠蛋白基因。下方为β-珠蛋白基因簇,位于11号染色体短臂互补链5.22-5.27 Mb区段,全长约50 kb,依次为ε、γ2、γ1、δ和β-珠蛋白基因。此外,α-珠蛋白基因簇上有两个假基因Ψζ和Ψα;β-珠蛋白基因簇上有1个假基因Ψβ。这10个珠蛋白基因在不同发育阶段表达,θ、μ、β和δ在成人血红细胞中表达,γ1和γ2在胎儿血红细胞中表达,ζ和ε在胚胎血红细胞中表达,而α1和α2在成人和胎儿血红细胞中均表达。

|
| 图 2 人α-珠蛋白和β-珠蛋白基因家族染色体定位 |
上述α和β-珠蛋白基因编码的血红蛋白氨基酸序列,均存放在国际蛋白质序列数据库UniProt中。利用该数据库提供的高级检索功能,可以快速有效地检索到这些蛋白质序列条目。具体检索步骤如下:
(1)点击UniProt数据库主页上方检索框右侧Advanced下拉式菜单,打开弹出式高级检索子窗口(图 3-A)。
(2)点击高级检索窗口最上方下拉式选择菜单中的All,选择Protein Name[DE],在其右侧的文本输入框中输入血红蛋白的英文Hemoglobin。
(3)点击第2个下拉式选择菜单中的All,选择基因名Gene Name[GN],在其右侧的文本输入框中输入血红蛋白的基因名缩写hb(不分大小写),并在后加通配符星号,即hb*。 (4)点击该选择菜单输入框右侧增加选择项符号“+”,弹出第3个选择菜单(图 3-B)。
(5)点击第3个选择菜单中的All,选择物种名Organism[OS],在其右侧输入Human,系统列出该数据库中与输入文本Human相关的所有物种,选择Human [9606]。9606为人在NCBI分类学数据库中的登录号。
(6)点击检索窗口右下侧检索按钮(图标为放大镜),提交检索策略,页面显示UniProt数据库中收录的所有人血红蛋白序列条目。
(7)点击页面左侧Reviewed图标,页面显示检索结果(图 3-C)。

|
| 图 3 利用UniProt 高级检索界面检索人血红蛋白9 个序列 条目 |
检索结果中列出已经通过人工审阅的9个人血红蛋白序列条目。UniProt数据库包括Swiss-Prot和TrEMBL两个子库,其中Swiss-Prot中的序列条目均已经通过人工审阅,而TrEMBL中的序列条目则是利用计算机对核酸序列数据库EMBL中的蛋白质编码序列翻译得到的,未经人工审阅。截止2015年3月,Swiss-Prot子库中的数据条目总数为547 599条,而TrEMBL子库中的数据条目总数为90 860 905条。显然,这两个子库的数据量差别极大。点击UniProt网站主页面下方UniProt data栏目下的Statistics图标,可以找到这两个子库的统计资料文档UniProt/Swiss-Prot statistics和UniProt/TrEMBL statistics,文档中有许多图表,详细叙述这两个子库的基本情况。
3 数据库序列相似性搜索 3.1 研究背景利用上述蛋白质序列数据库高级检索方法,可以快速高效地找到人血红蛋白基因家族9个成员所编码的蛋白质序列。近年来发现,除了运送氧气的血红蛋白和储存氧气的肌红蛋白外,人体中还有另外两种珠蛋白分子,一种为细胞红蛋白,或简称胞红蛋白(Cytoglobin),普遍存在于各种组织,可能具有氧储存、氧感受、一氧化氮运输、抗自由基等多种功能。另一种为神经红蛋白(Neuroglobin),多见于脑组织,因此也称脑红蛋白。胞红蛋白基因位于17号染色体长臂25区(17q25),编码190个氨基酸残基;脑红蛋白基因位于14号染色体长臂24区(14q24),编码151个氨基酸残基。X衍射晶体结构研究证明,这两种蛋白质分子的三维空间结构与血红蛋白、肌红蛋白具有相同折叠模式,同属珠蛋白家族(Globin family)。序列比对发现,两者与血红蛋白序列相似性均很低。胞红蛋白与血红蛋白α-亚基的相同位点共42个,约占22%;脑胞红蛋白与血红蛋白α-亚基的相同位点仅31个,不到20%。
3.2 搜索方法利用BLAST数据库相似性搜索,可以通过局部序列比对方法,从数据库中找到相似性较高的序列或序列片段。例如,以人血红蛋白α-亚基HBA_HUMAN为检测序列,可以从Swiss-Prot数据库中搜索到与其相似性较高的其它物种血红蛋白α-亚基序列。而对于脑红蛋白这样相似性很低的序列,则需要通过选择搜索程序、确定搜索数据库、限制搜索物种、设置适当的搜索参数,才能搜索到。具体步骤如下:
(1)打开NCBI BLAST服务器主页面,在常用BLAST选择区(Basic BLAST)中选择蛋白质BLAST(protein blast),将人血红蛋白α-亚基HBA_HUMAN序列粘贴到检测序列输入框。
(2)在数据库选择框(Database)中选择Swissprot protein sequence(swissprot),在物种选择框(Organism)中输入Human。
(3)在程序选择区选择位点特异迭代型BLAST(Position-specific Iterated BLAST),即PSI-BLAST。 (4)打开参数选择(Algorithm parameters)窗口,将错误率(Expected threshold)由缺省值10调为0.001。
(5)点击运行BLAST按钮递交作业,搜索结果得到11个珠蛋白分子。
(6)点击“运行第2次PSI-Blast”(Run PSI-Blast iteration 2 with max 50)按钮(Go),新一轮搜索结果中包括脑红蛋白(Neuroglobin,Siwss-Prot数据库登录号Q9NPG2.1)。
3.3 搜索结果搜索结果(图 4)显示,人12个珠蛋白均在搜索结果中,而与珠蛋白无关的其它序列则没有列在搜索结果中。也就是说,搜索结果既无假阳性(False positive)结果,也无假阴性(False negative)结果。

|
| 图 4 利用BLAST 从Swiss-Prot 数据库中搜索12 个珠蛋白 |
上述搜索过程说明,BLAST是一个功能强大的序列相似性数据库搜索系统。但要用好BLAST,必须对其基本算法有所了解,例如位置特异性迭代BLAST的原理、计分矩阵、错误率E值的选取等。
4 系统发生树构建 4.1 研究背景研究表明,人、小鼠和大鼠3种哺乳动物中,均有血红蛋白、肌红蛋白、胞红蛋白和脑红蛋白4类珠蛋白基因家族成员,其中肌红蛋白、胞红蛋白和脑红蛋白在这3个物种基因组中均为单拷贝基因,而血红蛋白α和β-两个亚家族均包含多个拷贝,在3个物种基因组中的数目、分布也不相同。美国宾夕法尼亚州立大学从事血红蛋白研究多年的哈迪森教授2012年发表的“血红蛋白及其基因的演化”综述中,对人和其它脊椎动物的血红蛋白起源、演化、表达和功能做了详细介绍[3]。图 5 是根据该论文中的插图改编的人、小鼠、大鼠3个物种基因组中α-和β-珠蛋白基因家族成员名称和在染色体上的排列次序。

|
| 图 5 人、小鼠、大鼠α-珠蛋白和β-珠蛋白基因家族 |
上述3个物种中,人类基因组的血红蛋白基因家族研究得比较清楚,而小鼠和大鼠血红蛋白的基因家族的大部分成员是根据基因组、转录组序列预测所得,尚无实验证据。表 3列出这3个物种中已经确定的37个成员。
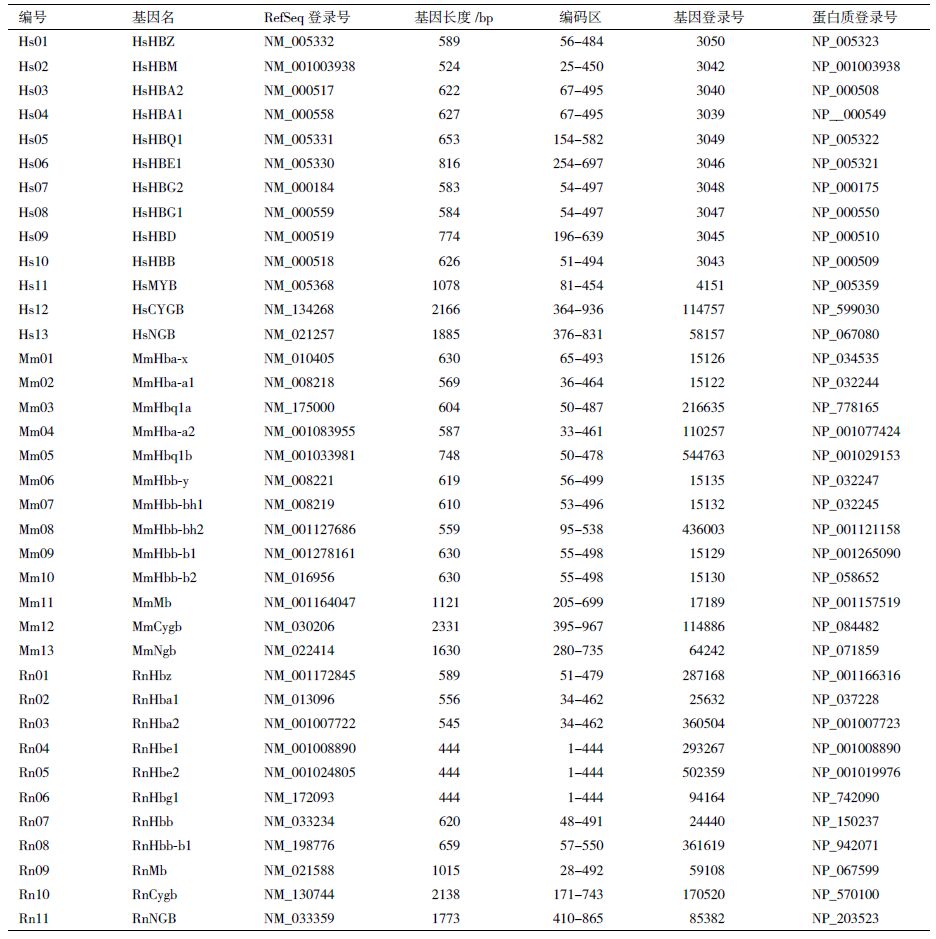
需要说明的是,小鼠脑红蛋白基因有两个剪接变体,RefSeq参考序列数据库中mRNA序列登录号为NM_022414和NM_001294308。NM_022414编码区长度453 bp,编码151个氨基酸;NM_001294308编码区长度465 bp,编码155个氨基酸。表中只列出其中一个NM_022414。小鼠β-珠蛋白家族成员MmHbb-b1和MmHbb-b2为单倍体型C57BL/-株系基因组中测得的序列,RefSeq参考序列数据库中mRNA序列登录号为NM_001278161和NM_016956。小鼠基因组计划测序样本所用的为融合体BALB/c和129Sv株系。小鼠基因组信息系统(MGI)中所列小鼠β-珠蛋白家族成员基因名为MmHbb-bs和MmHbb-bt,RefSeq mRAN登录号为NM_001201391和NM_008220,表中未予列出。
此外,大鼠基因组中α-珠蛋白家族共有7个成员[3],位于10号染色体15.468-15.508 Mb区段,长度约为40 kb;表 8中只收录已有转录数据的3个,即RnHbz(NM_013096)、RnHba1(NM_013096)和RnHba2(NM_001007722)。另4个尚无确切证据,也无确定的基因名,未在表中列出。这4个基因中,一个为α-珠蛋白,RefSeq数据库中mRNA序列登录号为 NM_001013853,大鼠基因组数据库RGD中暂定基因名为LOC287167;其它3个为θ-珠蛋白,尚无实验证据。大鼠基因组中,β-珠蛋白共有9个成员,位于1号染色体175.095-175.170 Mb区段,约75 kb(图 6),其中1个为假基因,4个为串联重复排列的α-珠蛋白,推测由近期发生的基因倍增机制产生。

|
| 图 6 大鼠基因组数据库RGD 中α-珠蛋白(A)和β-珠蛋白(B)基因家族信息 |
利用上述3个物种基因组中的血红蛋白及同一家族的肌红蛋白、胞红蛋白和脑红蛋白序列信息,可以构建分子系统发生树。系统发生树是以树状图表示不同物种之间系统发生关系的常用方法。达尔文“物种起源”一书中唯一的一幅插图,就是用树的形式表示物种多样性及其起源和演化。因此,系统发生树,有时也称“进化树”或“演化树”。其实,系统发生树不仅可以用来表示不同物种之间的亲缘关系和演化途径,也可以用来表示同一物种内部某个基因家族的不同成员之间的关系及演化。
利用MEGA软件包[4],可以构建人的珠蛋白基因家族12个成员系统发生树,所用序列为蛋白质序列,用全局比对程序ClustalW进行多序列比对,用GONNET蛋白质计分矩阵,空位罚分和其它参数均采用默认值。用邻接法(Neighbor-Joining)建树,采用差异位点比例(p-distance)为距离模型,选择自举法(Bootstrap)100次作为稳定性检验。
利用MEGA软件包中的邻接法(Neighbor-Joining)方法构建人、小鼠、大鼠3个物种珠蛋白基因家族37个成员系统发生树(图 8),所用序列为编码区核苷酸质序列。序列比对采用ClustalW Codon,即基于密码子的序列比对,比对过程中密码子3个核苷酸不打断,双序列和多序列比对的起始空位罚分均调为20,延伸空位罚分均调为2.0,以减少不必要的空位插入。建树过程中采用差异位点比例(p-distance)为序列差异模型,用转换加颠换(transition + transversion)为核苷酸替换模型,选择自举法(Bootstrap)100次作为稳定性检验。
4.3 结果分析图 7所示的系统发生树为基因树。结果表明,人的12个珠蛋白基因可以分为5个分支,其中α-珠蛋白亚家族包括4个成员,β-珠蛋白亚家族包括5个成员,而肌红蛋白、胞红蛋白和脑红蛋白各有1个成员。α-珠蛋白和β-珠蛋白有共同祖先,而肌红蛋白和胞红蛋白有共同祖先。α-珠蛋白亚家族4个成员中,α-珠蛋白和θ-珠蛋白之间的距离较近,而β-珠蛋白亚家族5个亚家族中,γ1-珠蛋白和γ2-珠蛋白的距离最近,其次为α-珠蛋白和δ-珠蛋白。

|
| 图 7 人珠蛋白家族12 个蛋白质序列系统发生树 |
图 8所示的系统发生树包括3个物种,每个物种均有多个基因,共37个基因。结果表明,37个基因总体可以分为5个分支,即α-珠蛋白、β-珠蛋白、肌红蛋白、胞红蛋白和脑红蛋白。3个物种的肌红蛋白、胞红蛋白和脑红蛋白各聚为一支;3个物种所有α-珠蛋白聚在一起,所有β-珠蛋白聚在一起。这一结果说明,这5类基因在3个物种形成以前就已经出现,即“先有基因、后有物种”。α-珠蛋白分为3支,第一支为ζ-珠蛋白,3个物种各有一个成员,即人的HsHBZ、小鼠的MmHba-x和大鼠的RnHbz;第二支又分两支,一支为α-珠蛋白,另一支为θ-珠蛋白。3个物种的α-珠蛋白各有两个成员,如人的HsHBA1和HsHBA2,θ-珠蛋白各有1个成员。可以推断,α-珠蛋白的两个成员是在灵长类和啮齿类分化以后通过基因倍增机制产生的,即“先有物种、后有基因”。β-珠蛋白基因簇在这3个物种的起源和演化留给读者自行分析。

|
| 图 8 人、小鼠、大鼠3 个物种珠蛋白家族系统发生树 |
基于蛋白质和核酸序列,我们已对人、小鼠和大鼠3个物种的血红蛋白进行了比较分析。下面,我们以斑头雁和灰雁为例,利用生物信息方法和结构分析软件,对血红蛋白的序列、结构和功能关系进行分析。
斑头雁在分类学上为鸟纲(Aves)、雁形目(Anseriformes)、鸭科(Anatidae)、雁属(Anser),拉丁文学名分别为Anser indicus,英文名为Bar-headed goose。斑头雁为典型的候鸟,夏季生活在我国西部青海湖,每年9月初往南迁徙,经过近两个月的长途跋涉,飞跃喜马拉雅山,大约10月中下旬飞抵印度平原过冬。每年春季开始又往北迁徙,飞回青海湖,周而复始,年年如此。灰雁(英文名为Grayleg goose,美国英语多用Greyleg goose)的拉丁文学名分别为Anser anser,与斑头雁同为鸭科、雁属,主要生活在印度平原[5]。我们知道,地球表面氧分压随海拔增高而降低,斑头雁飞跃的喜马拉雅山巅,氧分压不到平原地区的一半。斑头雁这种高空长度迁徙的能力,是否与其血红蛋白分子的特征有关,是一个值得研究的有趣问题。
1983年,英国剑桥分子医学研究实验室已故著名血红蛋白研究专家佩鲁茨(Max Perutz)在分子生物学和演化杂志(Molecular Biology and Evolution)创刊号上发表的题为“从蛋白质分子看物种的适应性”综述中指出,斑头雁和灰雁的血红蛋白氨基酸序列仅有4个位点差异,其中α-亚基的119位比较特殊[6]。斑头雁α-亚基该位点位序氨酸(A119Ala),而灰雁该位点为脯氨酸(A119Pro)。蛋白质三维空间结构分析表明,该位点与β-亚基第55位的亮氨酸(B55Leu)空间距离较近。我们知道,成熟的血红蛋白为四聚体,由两个α-亚基和两个β-亚基组成,各含一个血色素卟啉环,环中央的五价铁离子用于结合氧气。结合氧气和释放氧气过程中,血红蛋白四个亚基构象发生变化,并通过协同作用,提高结合氧气的效率。佩鲁茨指出,斑头雁α-亚基119位的丙氨酸侧链仅有一个甲基,与β-亚基55位亮氨酸侧链距离较远,有利于构象变化;而灰雁该位点侧链脯氨酸有3个甲基,与β-亚基55位亮氨酸侧链距离较近,不利于构象变化。这两种鸟类血红蛋白序列结构的差异,可能与其结合氧气的能力有关。20世纪90年代,北京大学生物系蛋白质结构功能研究组,用蛋白质分子晶体X-衍射的方法,分别测定了斑头雁和灰雁血红蛋白的结构,并进行了比较分析,证实了当年佩鲁茨的推测[7]。
5.2 研究方法利用蛋白质结构显示和模拟软件Swiss-PdbVie-wer[8],我们可以对已经测定的斑头雁和灰雁氧合血红蛋白的空间结构进行比较分析。具体操作步骤大体如下:
从蛋白质结构数据库PDB(http://www.rcsb.org/)下载斑头雁和灰雁氧合血红蛋白三维空间结构数据文件1A4F.pdb和1FAW.pdb。
(1)在Swiss-PdbViewer中打开灰雁血红蛋白数据文件1FAW.pdb,选择其中A和B两条链(即α和β-两个亚基),保存为新文件1FAWab.pdb。
(2)打开新保存的文件1FAWab.pdb,选择只显示主链模式;打开斑头雁血红蛋白数据文件1A4F.pdb,也选择只显示主链模式。
(3)利用该软件包中的结构叠合工具Magic Fit,可以发现,这两个蛋白质分子的结构总体十分相似。
(4)在控制面板中找到斑头雁α-亚基119位的丙氨酸和β-亚基55位亮氨酸,显示它们的侧链原子,测量它们之间的距离。
(5)在控制面板中找到灰雁α-亚基119位的脯氨酸和β-亚基55位亮氨酸,显示它们的侧链原子,测量它们之间的距离。
5.3 结果分析上述斑头雁和灰雁血红蛋白三维结构的比较分析表明,斑头雁氧合血红蛋白1A4F α-亚基119位丙氨酸侧链的β碳原子(CB)与β-亚基55位亮氨酸侧链末端的两个δ碳原子(CD1和CD2)距离均在4 Å以上,最近距离为4.56 Å;而灰雁该位点侧链脯氨酸γ碳原子与β-亚基55位亮氨酸侧链末端的一个碳原子距离为3.79 Å。这一差别很可能影响血红蛋在结合和释放氧气过程中构象发生变化,从而影响其结合氧气能力,造成这两种鸟类不同的生活习性。
图 9为利用PyMol分子结构显示软件绘制的分析结果。与Swiss-Pdbviewer相比,其图形显示和输出功能更强。

|
| 图 9 斑头雁(A)和灰雁(B)血红蛋白结构比较 |
以上我们以血红蛋白序列和结构为例,介绍“实用生物信息技术”课程教学种用到的几种生物信息方法。希望选修本课程的学生对本课程的教学有所了解,也希望对自学生物信息技术及其应用的读者有所启发。关于本课程的详细介绍和具体内容,读者可浏览本课程专用教学网站(http://abc.cbi.pku.edu.cn/),参阅笔者生物信息学简报(Briefings in Bioinformatics)相关文章[1]。
| [1] | Luo J. Teaching the ABCs of bioinformatics:a brief introduction to the Applied Bioinformatics Course[J]. Brief Bioinform, 2014, 15:1004-1013. |
| [2] | Liu X, Wu J, Wang J, et al. WebLab:a data-centric, knowledge-sharing bioinformatic platform[J]. Nucleic Acids Res, 2009, 37:W33-39. |
| [3] | Hardison RC. Evolution of hemoglobin and its genes[J]. Cold Spring Harb Perspect Med, 2012, 2:a011627. |
| [4] | Tamura K, Stecher G, Peterson D, et al. MEGA6:Molecular Evolutionary Genetics Analysis version 6. 0[J]. Mol Biol Evol, 2013, 30:2725-2729. |
| [5] | Jessen TH, Weber RE, Fermi G, et al. Adaptation of bird hemoglobi-ns to high altitudes:demonstration of molecular mechanism by prot-ein engineering[J]. Proc Natl Acad Sci USA, 1991, 88:6519-6522. |
| [6] | Perutz MF. Species adaptation in a protein molecule[J]. Mol Biol Evol, 1983, 1:1-28. |
| [7] | Zhang J, Hua Z, Tame JR, et al. The crystal structure of a high oxygen affinity species of haemoglobin[J]. J Mol Biol, 1996, 255:484-493. |
| [8] | Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer:a historical perspective[J]. Electrophoresis, 2009, 30:S162-173. |