



b. Leipzig University, Department of Systematic Botany and Functional Biodiversity Research, Johannisallee 21-23, 04103 Leipzig, Germany;
c. German Centre for Integrative Biodiversity Research(iDiv) Halle-Jena-Leipzig, Deutscher Platz 5e, 04103 Leipzig, Germany
Why are some regions more species-rich than others? This question has been at the focus of many studies over the past few decades (e.g. Antonelli and Sanmartín, 2011; Barthlott et al., 1996; Condamine et al., 2012; Favre et al., 2015; Graham et al., 2014; Tang et al., 2006). Besides the tropics, mountain systems have been found to be especially diverse, as reflected by the global distribution of species richness of vascular plants (Barthlott et al., 2005). Furthermore, a large proportion of terrestrial centres of diversity is associated with mountain systems (Fjeldså et al., 2012; Hughes and Atchison, 2015). Some have argued that orogenesis could have facilitated the establishment of high levels of biodiversity by providing a diversity of ecological opportunities on a remarkably small geographic scale (Dufour et al., 2006; Hoorn et al., 2013; Linder, 2008; Silvestro and Schnitzler, 2018). In addition, the "mountain geo-biodiversity hypothesis" by Mosbrugger et al. (2018), developed based on research in the QTP and for subtropical mountain systems, highlights the potential importance of the interaction between climatic and geological settings. This hypothesis suggests that, as a pre-requisite for diversification, surface uplift should have created full elevational zonation (from tropical to nival thermal belts), providing both refugia for the persistence of lineages during climate modifications as well as geographic barriers promoting allopatric speciation. Simultaneously to, or following, the uplift, mountains should have acted as "species pumps" (sensu Haffer, 1969) during the glacial cycles of the Pliocene-Pleistocene (Ehlers and Gibbard, 2004; Kaufman and Manley, 2004). Thus, a delay between the initial uplift and the biological response (diversification) might be observed, particularly in old mountain systems such as the Qinghai-Tibet Plateau (QTP) and others, as reviewed by Hughes and Atchison (2015).
For the QTP, the highest and largest plateau on Earth (Wang et al., 2014) resulting from at least 50 Myr of continuous orogenesis (Favre et al., 2015; Renner, 2016), several studies have uncovered shifts in net diversification rates of plant groups during the last few million years (Myr) (e.g. Ebersbach et al., 2016; Favre et al., 2016; Xing and Ree, 2017). These recent radiations have led to a few plant genera (e.g. Gentiana, Saxifraga, Corydalis, and Saussurea) being particularly well-represented in the alpine subnival belt flora of the Hengduanshan (Xu et al., 2014). More generally, the mountains surrounding the QTP (the Tianshan, the Himalayas, and the Hengduanshan) are particularly species-rich. Together with the relatively species-poor QTP interior, they harbour over 12, 000 vascular plant species (Zhang et al., 2016 and references therein). Favre et al. (2015) highlighted the importance of studying widespread groups, instead of only endemic taxa, to compare the evolutionary patterns between the region of interest and other mountain systems or ranges. Furthermore, the prevalence of biological interchange should be investigated, in order to better understand whether the QTP acted as a sink or a source area for plants. In the present study, we evaluate the "mountain-geo-biodiversity hypothesis" using the evolutionary and biogeographic history of the genus Allium L. (Amaryllidaceae), a taxon occurring in several mountain systems around the world.
Allium L. is one of the largest currently recognised monocotyledon genera, comprising at least 660 temperate and subtropical species (Choi and Oh, 2011; Govaerts et al., 2016; Gregory et al., 1998; Li et al., 2010; Xu and Kamelin, 2000), of which at least 111 occur within or surrounding the QTP. The genus is distributed across the Northern Hemisphere with a few exceptions (e.g. eastern and southern Africa), mainly in regions that are seasonally dry, such as the Irano-Turanian region (Choi and Oh, 2011; Govaerts et al., 2016; Xu and Kamelin, 2000). Its main centre of diversity is located between Southwest and Central Asia and the Mediterranean region, which is also supposed to be the major centre of diversification of Allium, besides a second one existing in North America (Choi and Oh, 2011). The genus is the only member of the monotypic tribe Allieae (Borkh.) Dumort., within the subfamily Allioideae Engl. (Amaryllidaceae J. St.-Hil.) (Chase et al., 2009). Allium is characterised by bulbs enclosed in membranous (sometimes fibrous) tunics, free or almost free tepals, and often a subgynobasic style (Friesen et al., 2006). Most species produce remarkable amounts of cysteine sulphoxides, causing the specific smell and taste of onion and garlic (Friesen et al., 2006). The taxonomy of Allium was revised in 2006 and 2010, based on morphological characters and rbcL sequence data (Friesen et al., 2006; Li et al., 2010). Nonetheless, a proliferation of synonyms and disagreement as to the subdivision of the genus still persists (Choi and Oh, 2011; Herden et al., 2016; Li et al., 2016; Sennikov and Seregin, 2015). For example, although there is a phylogenetic clustering of all five groups, the individual delineations of the subgenera Allium, Cepa (Mill.) Radić, Polyprason Radić, Reticulatobulbosa (Kamelin) N. Friesen, and Rhizirideum (G. Don ex Koch) Wendelbo are lacking phylogenetic support, or are shown to be polyphyletic (Li et al., 2010). On species level, the World Checklist of Allium (Govaerts et al., 2016) provides the most recent taxonomic account of the genus. Friesen et al. (2006) detected three distinct phylogenetic lineages in Allium, which were named: "First, Second, and Third Evolutionary Line" (EL1, EL2, EL3). It was proposed in 2011 to split the genus into three separate genera based upon these phylogenetic lineages (Banfi et al., 2011). The proposed names are Nectaroscordum Lindl. for EL1, Caloscordum Herb. for EL2, and Allium for EL3, but this new nomenclature has not yet been broadly adopted.
Here we present the most comprehensive molecular data set on Allium to date, including species consensus sequences that underwent multiple stages of quality control. Because the application of the correct taxonomy based upon a solid phylogenetic hypothesis is crucial to this study, we first ask (1) Are the current circumscriptions of the evolutionary lineages and subgenera supported by our phylogeny? Subsequently, we use the reconstructed phylogeny, combined with molecular dating, biogeographic analyses, and diversification rates estimation, to answer two further questions: (2) Did Allium species in the areas adjacent to the QTP evolve in situ, or did they disperse there, and if so, when and from where? and (3) Did shifts of diversification rates occur simultaneously in the QTP and other old mountains, as expected following the "mountain-geo-biodiversity hypothesis"?
2. Materials and methods 2.1. Species coverage and sequence alignmentsWe downloaded all 4842 Allium sequences available from GenBank (accessed in May 2015; Benson et al., 2013), as well as 23 outgroup sequences from the most closely related genera within Alloideae (Leucocoryne Lindl., Nothoscordum Kunth, Tristagma Poepp., and Tulbaghia L.) and one more distantly related species, Dichelostema multiflorum (Benth.) A. Heller (Chen et al., 2013). Sequences belonging to different molecular regions were aligned individually in Geneious 6.1.6 (Kearse et al., 2012). To take into account possible multiple accessions per species, we proceeded as follows: In a first step, all sequences from each marker with identical organism name (operational taxonomic unit: OTU) were grouped and automatically aligned with MAFFT ver. 7.221 (Katoh and Standley, 2013). In case of markers for which only partial sequences were provided (e.g. ITS, matK), we preferably used complete sequences as a guide to align the smaller fragments. If no complete sequence was available, we concatenated the sequences of the individual parts. The concatenated complete sequences were then aligned using the "auto" option. Fragments were added to these single species alignments with the option "–addfragments". Some sequences on GenBank were provided as reverse (R), reverse complement (RC) or as complement (C), meaning each marker could potentially be provided as one of four options (incl. the usual forward option F). We always used the "–adjustdirection" option in MAFFT, which tests for each sequence if the reverse complement fits the alignment better, thereby accounting for F and RC options. In addition, for all sequences with a distance larger than 0.1 to the other sequences (within species), we tested a reverse alignment. As this step again uses the "–adjustdirection" option, all four possible options (F, R, RC, C) were covered (F and RC from within MAFFT, and R and C through this additional step). In case this test failed and pairwise distances larger than 0.1 could not be resolved (e.g. reverse complementary fragments or mis-determinations), we excluded the data for this taxon and marker to account for mislabelled or otherwise erroneous data. For all remaining taxa (i.e. single marker alignments with within species distances below 0.1), a consensus sequence was generated for each marker per species, which was then used for the following steps.
If for a given marker at least 40 species were present, all consensus sequences of this marker were aligned in the same fashion as explained above. After a visual check of the individual alignments, they were concatenated and plastid and nuclear partitions were defined. Sequences with lengths shorter than 10 percent of the longest unaligned sequence were removed from the multi-species alignment (MSA) of the markers ITS, atpB-rbcL, rbcL, matK, psbA-trnH, rps16, trnL-trnF, and trnL-rpl32, of which the regions ITS, trnL-trnF, and trnL-rpl32 were represented by more than 50% of the taxa included. Our final MSA comprised 352 out of 1082 accepted Allium species (Govaerts et al., 2016), representing all accepted major lineages and subgenera.
2.2. Phylogenetic analyses and molecular datingAll phylogenetic analyses estimated parameters independently for the nuclear and plastid partitions. To test the monophyly of the ingroup for the subsequent molecular dating analysis and to identify potential problematic long branches, we conducted Maximum Likelihood (ML) and Bayesian Inference (BI) analyses. We produced a best scoring maximum likelihood tree (from 70 ML searches), based on the concatenated data and the partition table using RAxML v. 8.2.8 (Stamatakis, 2014) under the GTRGAMMA substitution model. Node support was calculated by a Bootstrap analysis with 350 iterations determined by the "bootstopping" algorithm (Pattengale et al., 2010) during the analysis. We performed Bayesian phylogenetic inference using a Metropolis-coupled Markov Chain Monte Carlo (MC3) approach as implemented in MrBayes v. 3.2.5 (Ronquist et al., 2012). Four independent analyses were run for 50 million generations with four incrementally heated chains, sampling from the posterior probability distribution every 5000th generation. In addition, we used the model-jumping approach to sample all possible 203 substitution models according to their posterior probability (Ronquist et al., 2012) in combination with a gamma model of rate heterogeneity. After confirming that the four independent runs converged on the same solution space judged by effective sample size (ESS) values well above 200 and homogeneous "white noise"-like traces of both the individual runs and the combined log, a majority-rule consensus tree was constructed from the combined runs (burn-in: 36%, as indicated by ESS values). Furthermore, we compared the reconstructed phylogeny of a simple concatenated non-partitioned matrix with the one of a partitioned (plastid/nuclear) matrix to visualize potential alternative topologies in Allium (Appendix 1). Major topological incongruencies only appeared in the Third Evolutionary Line, and did not affect our analyses. A likelihood-ratio test rejected the null hypothesis of clocklike evolution of the analysed sequences (p < 0.001).
Divergence times were therefore estimated under an uncorrelated lognormal relaxed clock, using BEAST v.1.8.2 (Drummond et al., 2012), based on a data set from which we removed the markers with the highest proportions of missing data (atpB-rbcL, rbcL, matK, psbA-trnH, rps16), as these showed to impede MCMC convergence in preliminary analyses. In the absence of known fossils of Allium or closely related genera (Smith, 2013), we used two secondary calibration approaches on Allium's root node (i.e. crown node of Allioideae) from a recently published chronogram of Amaryllidaceae (Chen et al., 2013), defining (A) a rather narrow exponential prior distribution around their suggested age of 37 Myr (spanning from 37.0 at the 2.5% quantile to 37.4 at the 97.5% quantile with setting the offset to 37.0 and the mean to 0.1) and (B) a normal prior distribution spanning the age range Chen et al.(2013) found (27.8-44.5 Myr), with a mean of 37 Myr and a standard deviation of 4.5 Myr to tailor the distribution's 2.5% and 97.5% quantiles to 28.2 Myr and 45.8 Myr, respectively. Both analyses were run four times, sampling every 10, 000th of the 50 million MCMC generations in (A) and 100 million MCMC generations in (B). ESS values well above 200 for the combined log after removing varying proportions (10-25%) of burn-in from individual runs confirmed that convergence was reached. Therefore, we combined the tree files of the individual runs each in (A) and (B) and afterwards produced a maximum clade credibility (MCC) tree with common ancestor heights as described by Heled and Bouckaert (2013) using LogCombiner and TreeAnnotator (both v. 1.8.3)(Bouckaert et al., 2014). The alignments and consensus/MCC trees are available from the corresponding author upon request.
2.3. Diversification ratesWe used BAMM (Rabosky, 2014), including the R package BAMMtools (Rabosky et al., 2014), to assess the diversification rate heterogeneity within Allium. We reduced the MCC tree to only one OTU per species (removing all subspecies and varieties), and specified lineage-specific sampling fractions to account for incomplete taxon sampling. We ran four MCMC chains (15 million generations, every 5000th generation sampled, 10% burn-in) to identify distinct configurations of rate shifts. However, some limitations of BAMM have recently been identified (Moore et al., 2016), including strong prior sensitivity and potentially unreliable rate estimates (but see Rabosky et al., 2017). Thus, we additionally used BayesRate (v.1.6.5, Silvestro et al., 2011) to evaluate the different diversification scenarios. Importantly, BayesRate employs a likelihood function (Nee et al., 1994) different from the one implemented in BAMM (Maddison et al., 2007), and thus provides an independent evaluation of the diversification model (number of rate shifts) and the associated rates. We compared the most likely scenario identified by BAMM with both simpler and more complex models of diversification (i.e. fewer or more rate shifts) via thermodynamic integration and estimated model parameters for the best-fit model, again accounting for incomplete taxon sampling.
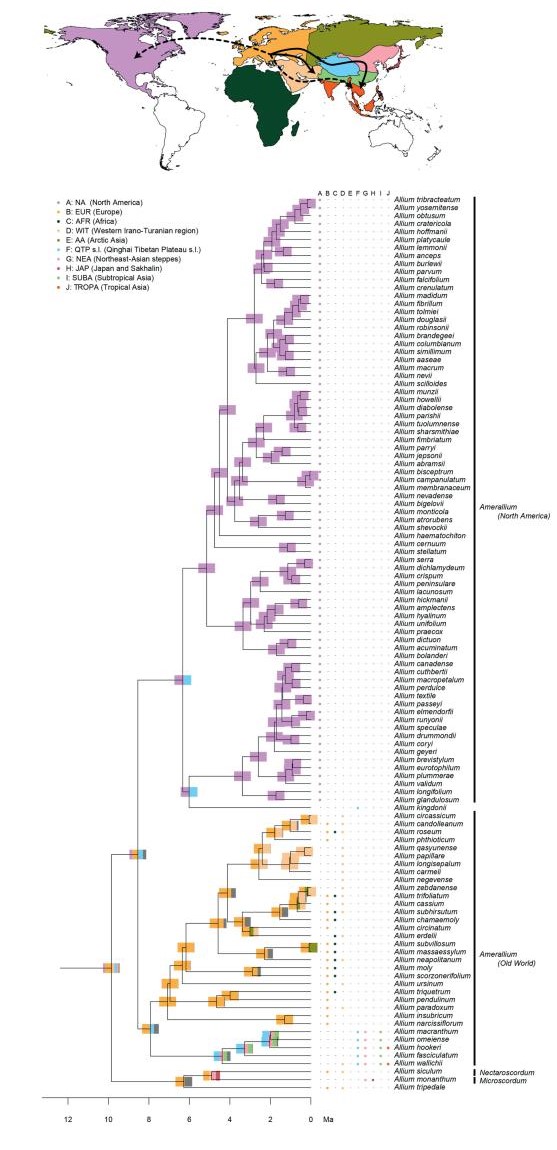
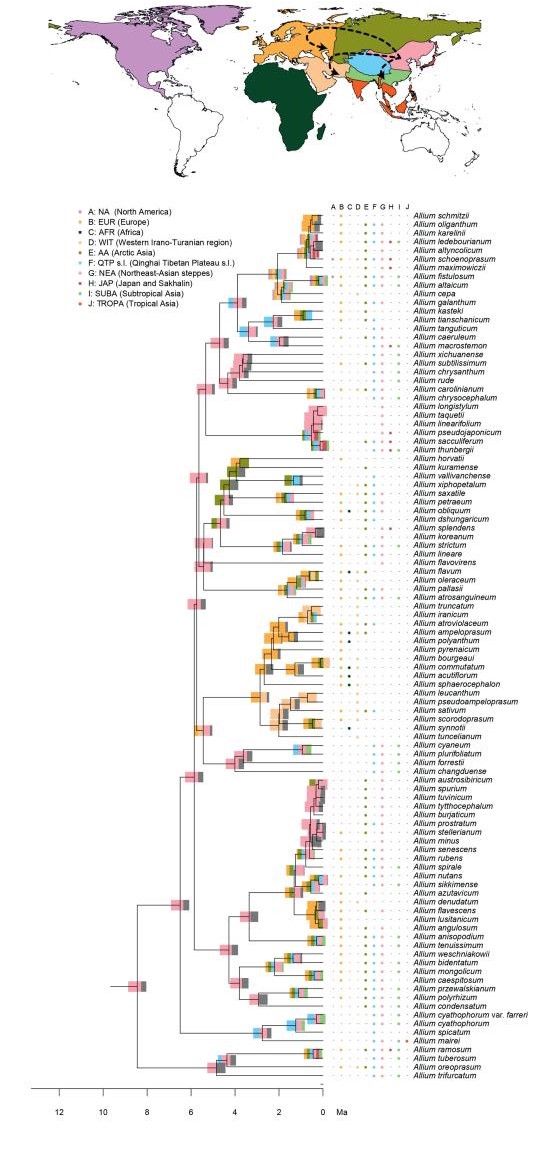
2.4. Biogeographic analysisTo estimate ancestral geographic distributions we performed biogeographic analyses using the R (R Core Team, 2015) package BioGeoBEARS (Matzke, 2014) under the DEC model (Ree, 2005; Ree and Smith, 2008) and a likelihood version of the DIVA (Ronquist, 1997) model with or without the inclusion of a jump dispersal (i.e. founder event) parameter (Matzke, 2014). The maximum combination of areas parameter was set to 7. We did not set time strata, manual dispersal multipliers, and area restrictions, to avoid sharp temporal borders on a secondarily calibrated reconstruction. The best model for the data set among those four was chosen based on AIC values. Eleven geographic areas were defined with a more fine-scale delineation for the QTP and the regions neighbouring it. These were delineated using their ecological attributes (climate, topography, etc.), while other regions (continental scale) were coded according to their interconnectivity. In detail, the continental regions were coded as follows: A: NA: North America, B: EUR: Europe s.l. (eastern borders being the Caucasus mountains and Ural mountains), and C: AFR: Africa. The Asian continent was delimited as follows: D: WIT: Western Irano-Turanian region, E: AA: Arctic Asia, (East of the Ural and North of the southern Siberian mountain chains), F: QTP: QTP s.l. (Qinghai-Tibet Plateau, Tian Shan, Hengduanshan, and the Himalayas). G: NEA: Northeast-Asian steppes (surrounded by Area AA in the North, QTP in the West; the border follows the Han River and further the Yangtze River in the South), H: JAP: Japan and Sakhalin. The rest of the Asian land (surrounded by region WIT in the West, QTP in the North, the Pacific Ocean in the East, and the Wallace Line in the South) is separated by both, seasonality and elevation, resulting in I: SUBA: Subtropical Asia and J: TROPA: Tropical Asia (Appendix 2).
Extant species' distributions were based on more than 2.7 million individual occurrence points from the literature and herbarium vouchers. In taxa for which this procedure did not provide data, we added more coarse distribution information from the World Checklist of selected Plant Families (Govaerts et al., 2016). In order to match the taxonomic units in the distribution data to those in the chronogram, we used the synonymy list from Govaerts et al. (2016) to merge entries in both, the occurrence data and in the chronogram. Subsequently, the gathered distributional data was transformed to presence/absence data of the eleven defined geographic areas. To perform the biogeographic analyses we removed the species for which we could not obtain any distribution data, as well as subspecific taxa from the tree. Furthermore, Allium comprises a notable number of cultivated species (Mabberley, 2008). As the distribution of those species has been largely influenced by humans, we preferred coding their native areas of distribution.
3. Results 3.1. Phylogenetic analysis, molecular datingWe compiled a data set of 352 OTUs (including 18 subspecies, 18 varieties, one forma, and four undetermined specimens) with 12306 aligned base pairs from eight markers: ITS (1037 bp), atpB-rbcL (1008 bp), matK (1595 bp), psbA-trnH (882 bp), rbcL (3222 bp), rps16 (1327 bp), trnL-trnF (1214 bp), trnL-rpl32 (2021 bp). Our phylogenetic reconstructions are overall similar to the reconstructions by Friesen et al. (2006) using ITS, and Li et al.(2010) with ITS and rps16 (Fig. 1). Partition-dependent topological variations are present, yet none affects the arrangement of major lineages. All three previously recognized evolutionary lines are present and supported in our phylogeny (Fig. 1; posterior probabilities: 0.97, 1.00, and 1.00 respectively, bootstrap values: 75, 100, and 100), and our reconstruction of EL3 (Friesen et al., 2006) is largely similar to previous topologies. Furthermore, we observed two taxa (Allium condensatum Turcz. and Allium kingdonii Stearn), which were placed differently in our phylogeny, when compared to Friesen et al. (2006). Yet, consistent with the results of Huang et al.(2014), A. kingdonii is nested within subgenus Amerallium Traub., as sister to the clade of American Amerallium. Our phylogenies support the previously described subgenera Nectaroscordum (Lindl.) Asch. & Graebn., Microscordum (Maxim.) N. Friesen, Amerallium, Caloscordum (Herb.) R. M. Fritsch, Anguinum (G. Don ex Koch) N. Friesen, Poryphyroprason (Ekberg) R. M. Fritsch, Vvedemskya (Kamelin) R. M. Fritsch, Melanocrommyum (Webb. & Berth.) Rouy., Butomissa (Salisb.) N. Friesen, Cyathophora (R. M. Fritsch) R. M. Fritsch, and Rhizirideum (G. Don ex Koch) Wendelsbo s.s. The subgenera Allium, Reticulatobulbosa, Polyprason, and Cepa, and the above-mentioned A. condensatum (formerly Rhizirideum) are nested within each other (Fig. 1).

|
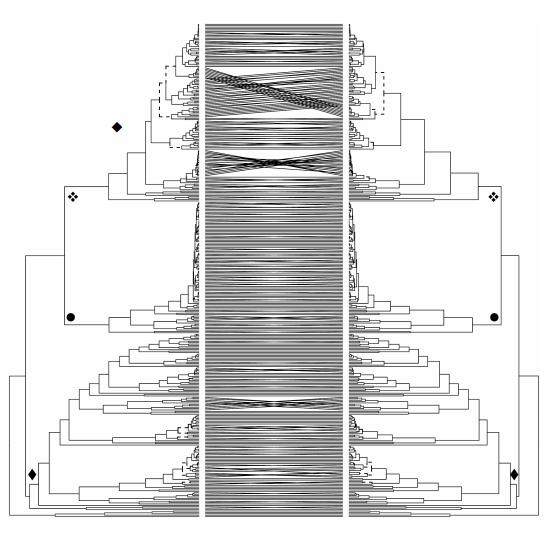
| Fig. 1 The maximum clade credibility (MCC) tree of Allium as reconstructed by BEAST. A. Monophyletic subgenera are collapsed. The subgenera Allium, Cepa, Reticulatobulbosa, and Polyprason do not form monophyletic non-exclusive groups, hence this clade is displayed uncollapsed. Support values above pp = 0.96 are shown by thicker lines. Evolutionary lines are represented by symbols (1st = ◆, 2nd = ●, 3rd = ❖). The classification (Evolutionary Lines) refers to Friesen et al. (2006). B. Full overview of the tree, highlighting the First (light grey), the Second (black) and the Third (dark grey) Evolutionary Lines. |
The crown node of Allium was estimated to be at least 12.8 (±1.6) Myr old. The crown of EL1 was reconstructed to be at least 10.2 (±1.4) Myr old. EL2 was estimated to have evolved between 10.0 (±1.4) and 6.6 (±1.4) Ma, and EL3 between 10.0 (±1.4) and 8.7 (±1.2) Ma (Fig. 2).

|
| Fig. 2 Diversification dynamics in Allium. The tree shows the best-fit model of diversification by BayesRate (A), identical colours indicate that speciation and extinction rates were linked between clades. Marginal posterior densities of the speciation, extinction, and net diversification rates (B) for the clades defined in (A). Ⅰ: a part of subgenus Amerallium in EL1◆. Ⅱ: the subgenera Allium, Cepa, Polyprason, and Reticulatobulbosa in EL3❖. Ⅲ: major parts of subgenus Melanocrommyum in EL2●. |
BAMM identified three shifts of net diversification rates: the North American part of subgenus Amerallium (Ⅰ), a clade comprising the Eurasian subgenera Allium, Cepa, Polyprason, and Reticulatobulbosa (Ⅱ), and the QTP-centred section of the Central Asian subgenus Melanocrommyum (Ⅲ) (Fig. 2). The five most credible scenarios (15.8%, 8.8%, 5.9%, 4.7%, and 4.7% of samples, respectively) all reconstructed rate shifts in the same groups (Ⅰ, Ⅱ, Ⅲ, cf. Fig. 2A), yet in groups Ⅱ and Ⅲ, the location of the rate shift is not constant among the scenarios, while the rate shift related to group Ⅰ occurs at the same branch among all five most credible scenarios. The sixth most credible scenario did not involve any significant rate shift (4.1% of samples), followed by two scenarios with three rate shifts similar in position to the five most credible scenarios (2.3% and 2.0% of samples, respectively). All other scenarios received probabilities below 2.0%. The data can be obtained from the corresponding author upon request.
The analyses with BayesRate confirmed the presence of three diversification rate shifts within Allium (Table 1). The highest marginal likelihood was found for a model with three different rates assigned to clades Ⅰ, Ⅱ + Ⅲ, and the remaining lineages, respectively (Fig. 2). The posterior distributions of the diversification rates (Fig. 2B) show an almost two-fold increase in the diversification rate (r) between the QTP-centred section of subgenus Melanocrommyum (Ⅲ; r = 1.371, 95% HPD [0.932; 1.897]) and clades Ⅰ and Ⅱ (r = 0.771, 95% HPD [0.605; 0.942]), while the posterior rate estimate for the remaining clades is substantially lower (r = 0.365, 95% HPD [0.220; 0.528]). These differences are mainly due to variation in speciation rates across Allium, while the extinction rate shows a much less pronounced distinction between clades (Fig. 2B).
| Model | logML | BF |
| Ⅰ|(Ⅱ + Ⅲ)|Rest | -438.681 | 0 |
| (Ⅰ + Ⅱ + Ⅲ)|Rest | -440.264 | 3.166 |
| Ⅰ|(Ⅱ + Ⅲ + Rest) | -443.875 | 10.388 |
| All linked | -448.748 | 20.134 |
| All unlinked | -452.78 | 28.198 |
Model evaluation identified the models without founder events as most suitable models (weighted ratio: 2.73), and most likely with the DEC model. However, all models achieved overall similar likelihoods and favoured consistent scenarios. The geographic origin of Allium could not be reconstructed with high confidence (eight areas, each with less than 50% probability, Appendix 2). EL1 was reconstructed to be of European (EUR) origin (less than 50% probability). Within subgenus Amerallium, the North American taxa were reconstructed to have originated in North America (NA, Appendix 2), while the Asian, African, and European taxa were estimated to be of European (EUR) origin ( < 50% to >98%, Appendix 2). The ancestral area of EL2 could not be resolved (eight areas, each with less than 50% probability, Appendix 2). Within this lineage, a similar pattern was reconstructed for the subgenera Caloscordum (four areas of overall similar probability), and Anguinum (eight areas of potential origin). The ancestral area of the subgenera Melanocrommyum, Poryphyroprason, and Vvedemskya were reconstructed to be of Arctic Asian (AA) origin, and within Melanocrommyum, major parts of its diversification were reconstructed to have occurred in the Western Irano-Turanian (WIT) and the QTP region (Appendix 2). EL3, and a major part of its internal lineages, was reconstructed to be of Northeast Asian (NEA) origin (Appendix 2). In contrast to EL2, the ancestral area of extant taxa distributed in the QTP was not reconstructed as being the QTP itself.
4. DiscussionWe performed phylogenetic, biogeographic, and diversification rate analyses using the widespread genus Allium as a model group to test the "mountain geo-biodiversity hypothesis". By gathering a large data set of available sequence data, combined with two data sets on distributional data, we could reveal new information related to the taxonomy within the genus by providing phylogenetic support to the monophyly of several subgenera (Fig. 1). Furthermore, we could identify a dual biogeographic pattern for Allium species occurring within the QTP region: while multiple taxa of one lineage seem to have immigrated from other areas (e.g. the Arctic and Northeast Asia) into the QTP and adjacent regions (e.g. species of EL3), the QTP acted more as a source area in a clade of subgenus Melanocrommyum (Appendix 2). Finally, although we recognize three diversification rate shifts occurring either in Asia or North America, the conditions subtending the "mountain-geo-biodiversity hypothesis" were only partially verified.
4.1. Taxonomy and the consensus approachTo arrive at solid biogeographical interpretations, one should ideally rely on a well-supported phylogenetic reconstruction, including a reasonable sampling across taxonomic units (e.g. subgenera, sections) and geographic areas. In our data set, all major lineages of Allium were included across the entire distribution range of the genus. Nevertheless, some clades and/or regions were better represented than others. For example, species coverage for areas which had already been targeted by phylogenetic studies, such as Korea and northeastern China (Choi and Oh, 2011), was higher than for regions and subgenera not yet revised in detail (e.g., the Americas). In general, our results are in line with the last revision of Allium (including 15 subgenera, organised into three evolutionary lines; Friesen et al., 2006), and with more recent studies targeting some of these subgenera, such as Anguinum and Cyathophora (Choi and Oh, 2011; Herden et al., 2016; Li et al., 2016). Nevertheless, several subgenera (Cepa, Reticulatobulbosa, Polyprason, and Allium) did not receive sufficient phylogenetic support in our analyses, or were placed in a polytomy. In addition, other subgenera, such as Microscordum, were only represented by one species, precluding any conclusions regarding their monophyly. Further phylogenetic studies on Allium should therefore primarily aim at resolving these phylogenetic uncertainties. Nevertheless, our automated approach was successful in recovering all clades previously described as monophyletic (Fig. 1). This result supports the argument of Banfi et al. (2011) for a nomenclatural revision, encouraging the split of Allium into three genera, namely Nectaroscordum, Caloscordum, and Allium. In the absence of major discrepancies between our phylogenies and Friesen et al.'s (2006) recent classification of Allium, we consider our data set suitable to reconstruct the evolutionary history of Allium.
4.2. Spatio-temporal dynamics of Allium evolutionAllium probably originated between 11.2 and 14.4 Ma (Appendix 2) in a yet undetermined area, potentially in a combination of several areas. Because there is no satisfactory paleobotanical data for Allium and closely related taxa, as is often the case for temperate herbaceous plants (mostly) pollinated by insects, we were unable to perform a thorough quality check on the temporal estimates. Yet, our time estimates are in line with other molecular dating approaches targeting larger clades including Allium and other genera within Amaryllidaceae (Conrad, 2008; Magallón et al., 2015). However, our estimates do not support the hypotheses of Li et al. (2010), who postulated that the First Evolutionary Line (EL1) originated around the Cretaceous-Paleogene boundary, to fit with possible vicariance events. Moreover, despite the lack of certainty regarding the geographical origin of the genus, we could recover the likely origins of major lineages and subgenera within the three evolutionary lines within Allium (Appendix 2).
It is likely that Europe was the centre of origin of the common ancestor of EL1, during the Late Miocene. Subgenus Amerallium dispersed from Europe first to North America (where it later diversified), and later on, from Europe to the Western Irano-Turanian region (potentially multiple times). However, our phylogenetic placement of the QTP-related A. kingdonii (different when compared to previous phylogenetic work, e.g. Friesen et al., 2006) obscures the precise route for the dispersal towards North America. Hence, our results do not contradict the spatial findings of Li et al. (2010), who proposed dispersal from eastern Asia to western North America. Similarly, subgenus Microscordum dispersed from Europe to the QTP, eastern and Tropical Asia. In contrast, ancestral area reconstructions lacked support for the origin and dispersal routes of EL2. Subgenera Melanocrommyum, Poryphyroprason, and Vvedemskya probably originated in an area including Europe and/or northern Asia (AA) around the Miocene/Pliocene boundary. Hence, like in EL1, Europe and/or the Euro-Siberian region seems to constitute the centre of origin, rather than solely eastern Asia, as claimed by Li et al. (2010). During the Early Pliocene, subgenus Melanocrommyum dispersed to the Western Irano-Turanian region, and later on back to northern Asia, possibly via the QTP region, which was associated with an increase in the rate of speciation (see below). Clearly, both the Irano-Turanian and the QTP region acted successively as a sink and a source area for dispersal. In fact, the former region likely was a source area for many drought-tolerant taxa (Manafzadeh et al., 2016). These findings are, to a large extent, in agreement with the hypotheses by Li et al. (2010), who also recognized a relatively recent centre of diversification in Central Asia for Melanocrommyum. Finally, we could locate the origin of EL3 in northeastern Asia (area NEA, including, among others, Mongolia), by the end of the Miocene. From there, Eurasian subgenus Butomissa and its sister clade (comprising Allium, Cepa, Cyathophora, Reticulatobulbosa, and Rhizirideum) dispersed mostly towards northern Asia (area AA) and Europe, where they diversified and are still abundant today. From northeastern Asia, these subgenera also repeatedly colonised more southern areas as well as the QTP. Overall, we have uncovered a highly complex biogeographic history, with multiple dispersal events between Eurasian areas. In fact, only the colonisation of North America and South or Southeast Asia appears to have been unidirectional.
4.3. Shifts in diversification rates and the "mountain-geo-biodiversity hypothesis"As postulated by the "mountain-geo-biodiversity hypothesis" (Mosbrugger et al., 2018), diversification in the region of the QTP may have required several conditions to be initiated, including the development of a full elevational zonation and climate oscillations promoting a "species pump" effect. In this region, the start of the uplift largely predated climate oscillations (by several Myr), suggesting that a delay of biological (diversification) processes might have occurred with respect to the uplift's start. Such a delay was indeed observed for some plant genera (Saxifraga, Ebersbach et al., 2016, and Gao et al., 2015; Gentiana, Favre et al., 2016) that were shown to have been present in the QTP for an extended period of time before the onset of their radiation. In Allium, the diversification of subgenus Melanocrommyum is generally consistent with the "mountain-geo-biodiversity hypothesis". The QTP-centred clade shows the highest rate of diversification, with the start of the radiation (crown node age estimate 2.34 Ma, 95% HPD [3.04:1.56]) coinciding with the onset of the Pleistocene climatic fluctuations. However, it should be noted that our biogeographic reconstructions suggest that the clade was probably not present in the QTP before that time. Thus, the higher rate of diversification could be either the result of a "species pump" effect following climatic oscillations, or be due to an ecological radiation following colonisation from the Western Irano-Turanian region. In addition, it should be noted that the exact position of this rate shift was not stable among the most credible scenarios. As a result, we cannot ascertain whether the increased speciation rate is associated with only the QTP and Arctic Asia, or extends to the Western Irano-Turanian region (Appendix 2). The other two diversification rate shifts (in the North American clade of subgenus Amerallium and the clade encompassing the subgenera Allium, Cepa, Cyathophora, Reticulatobulbosa, and Rhizirideum in northeastern Asia) predate the Pleistocene climatic oscillations by several Myr, both starting in the Late Miocene. Therefore, Allium does not fully support the "mountain-geo-biodiversity hypothesis". Rather, the species richness of Allium in the Northern Hemisphere (including its mountain systems) might have been triggered by global cooling facilitating a complex pattern of biological interchange between Eurasian regions, potentially aiding allopatric speciation and ultimately diversification.
The "mountain-geo-biodiversity hypothesis" also postulates that mountains should buffer against extinction during climate modifications by providing refugia and suitable habitats within a short distance, as already suggested by Hoorn et al. (2013). However, we do not find much evidence for this in our analysis. Extinction rates were found to be relatively low across Allium, regardless of whether a subclade occurred predominantly in mountain systems (e.g. subgenus Melanocrommyum in the QTP and the Irano-Turanian region), in the Euro-Siberian lowlands (subgenera Allium, Cepa, Cyathophora, Reticulatobulbosa, and Rhizirideum), or across North America (e.g. subgenus Amerallium). Consequently, increased speciation rates account for most of the higher net diversification rates. Similar findings were reported in Ericaceae by Schwery et al. (2015). However, in contrast, bellflowers (Lagomarsino et al., 2016), and the Paleo-Patagonian flora (Palazzesi et al., 2014) indicate potentially higher extinction rates in lowland areas. Hence, we need to gather further insights into other widespread taxa to test whether extinction rates in the region of the QTP were lower in comparison to those in the Eurasian lowland, and attest the role of the QTP region as buffer against extinction.
5. ConclusionsOur results show how the interaction among plant taxa, geology, and climate contributes to patterns of plant diversity in the region of the QTP (including the Hengduan Mountains) and beyond. Some of the results presented here indeed provide support for the "mountain-geo-biodiversity hypothesis". However, other processes such as ecological radiations, where species diverge as populations adapt to novel habitats in topographically complex regions, cannot be ruled out. In fact, we suggest that most likely both allopatric speciation (via the "species pump" effect) and ecological divergence act together in driving plant diversification in mountain regions. Both processes might lead to different ecological patterns, with speciation driven by climatic oscillations generally resulting in little ecological differences, whereas ecological radiations should show a high degree of ecological niche divergence between closely related species. Hence, further studies, especially concerning the ecological niche, are needed to evaluate the relative importance of climate-driven and ecological diversification processes.
AcknowledgementsAll analyses were performed on the High-Performance Computing Cluster EVE, a joint effort by the Helmholtz Centre for Environmental Research (UFZ) and the German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig. We thank the German Research Foundation for funding of staff (project no. MU 2934/2-1). We also gratefully acknowledge the support of the German Centre for Integrative Biodiversity Research (iDiv) HalleJena-Leipzig funded by the German Research Foundation (FZT 118) for funding of lab expenses.
Appendix 1Comparison of phylogenies (uncalibrated ultrametric trees) of Allium (black) and outgroups (grey) on the base of a concatenated nonpartitioned data set (left) and a partitioned data set (right, nuclear/plastid). The evolutionary lines one to three (EL 1-3) are labelled by symbols (◆: First, ●: Second, ❖: Third Evolutionary Line).
|
Ancestral area reconstructions in Allium, divided into three parts (Fig. 1: Evolutionary Line (EL) 1, Fig. 2: EL2, and Fig. 3: EL3). Node bars illustrate the percentage of an area being reconstructed as ancestral area, represented by colours from the map. The column between the tree and the species names displays the extant (non-introduced) range. Arrows on the map show reconstructed dispersal routes (lines) and potential dispersal routes (dotted lines). Subgenera are only given for EL1 and EL2, as the majority of subgenera described in EL3 are not monophyletic.
|

|
|
Antonelli A., Sanmartín I., 2011. Why are there so many plant species in the neotropics?. Taxon, 60: 403-414. |
Banfi E., Galasso G., Soldano A., 2011. Notes on systematics and taxonomy for the Italian vascular flora.2. Nat. Hist. Sci., 152: 85-106. DOI:10.4081/nhs.2011.85 |
Barthlott W., Lauer W., Placke A., 1996. Global distribution of species diversity in vascular plants:towards a world map of phytodiversity (Globale Verteilung der Artenvielfalt Höherer P flanzen:Vorarbeiten zu einer Weltkarte der Phytodiversität). Erdkunde: 317-327. |
Barthlott W., Mutke J., Rafiqpoor D., Kier G., Kreft H., 2005. Global centers of vascular plant diversity. Nova Acta Leopoldina NF, 92(342): 61-83. |
Benson D.A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W., 2013. GenBank. Nucleic Acids Res., 41: D36-D42. DOI:10.1093/nar/gks1035 |
Bouckaert R., Heled J., Kuhnert D., Vaughan T., Wu C.H., Xie D., Suchard M.A., Rambaut A., Drummond A.J., 2014. BEAST 2:a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol., 10. |
Chase M.W., Reveal J.L., Fay M.F., 2009. A subfamilial classification for the expanded asparagalean families Amaryllidaceae, Asparagaceae and Xanthorrhoeaceae. Bot. J. Linn. Soc., 161: 132-136. DOI:10.1111/(ISSN)1095-8339 |
Chen S., Kim D.-K., Chase M.W., Kim J.-H., 2013. Networks in a large-scale phylogenetic analysis:reconstructing evolutionary history of Asparagales (Lilianae) based on four plastid genes. PLoS ONE, 8: e59472. DOI:10.1371/journal.pone.0059472 |
Choi H.J., Oh B.U., 2011. A partial revision of Allium (Amaryllidaceae) in Korea and north-eastern China. Bot. J. Linn. Soc., 167: 153-211. DOI:10.1111/boj.2011.167.issue-2 |
Condamine F.L., Sperling F.A., Wahlberg N., Rasplus J.Y., Kergoat G.J., 2012. What causes latitudinal gradients in species diversity? Evolutionary processes and ecological constraints on swallowtail biodiversity. Ecol. Lett., 15: 267-277. DOI:10.1111/ele.2012.15.issue-3 |
Conrad, F., 2008. Molecular Systematics, Biogeography and Dating of the Tribe Haemantheae (Amaryllidaceae) and the Phylogeography of Clivia. University of Cape Town. https: //open. uct. ac. za/handle/11427/6243
|
Drummond A.J., Suchard M.A., Xie D., Rambaut A., 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol., 29: 1969-1973. DOI:10.1093/molbev/mss075 |
Dufour A., Gadallah F., Wagner H.H., Guisan A., Buttler A., 2006. Plant species richness and environmental heterogeneity in a mountain landscape:effects of variability and spatial configuration. Ecography, 29: 573-584. DOI:10.1111/eco.2006.29.issue-4 |
Ebersbach J., Muellner-Riehl A.N., Michalak I., Tkach N., Hoffmann M.H., Röser M., Sun H., Favre A., 2016. In and out of the Qinghai eTibet Plateau:divergence time estimation and historical biogeography of the large arcticalpine genus Saxifraga L. J. Biogeogr., 44: 900-910. |
Ehlers, J., Gibbard, P. L., 2004. Quaternary glaciations -extent and chronology: part Ⅰ: Europe, vol. 2. Elsevier.
|
Favre A., Michalak I., Chen C.-H., Wang J.-C., Pringle J.S., Matuszak S., Sun H., Yuan Y.-M., Struwe L., Muellner-Riehl A.N., 2016. Out-of-Tibet:the spatiotemporal evolution of Gentiana (Gentianaceae). J. Biogeogr., 43: 1967-1978. DOI:10.1111/jbi.12840 |
Favre A., Päckert M., Pauls S.U., Jähnig S.C., Uhl D., Michalak I., MuellnerRiehl A.N., 2015. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol. Rev., 90: 236-253. |
Fjeldså J., Bowie R.C.K., Rahbek C., 2012. The role of mountains in the diversification of birds. Annu. Rev. Ecol. Evol. Syst., 43: 249-265. DOI:10.1146/annurev-ecolsys-102710-145113 |
Friesen N., Fritsch R.M., Blattner F.R., 2006. Phylogeny and new intrageneric classification of Allium (Alliaceae) based on nuclear ribosomal DNA ITS sequences. Aliso, 22: 372-395. DOI:10.5642/aliso |
Gao Q.B., Li Y.H., Gornall R.J., Zhang Z.X., Zhang F.Q., Xing R., Fu P.C., Wang J.L., Liu H.R., Tian Z.Z., Chen S.L., 2015. Phylogeny and speciation in Saxifraga sect.Ciliatae (Saxifragaceae):evidence from psbA-trnH, trnL-F and ITS sequences. Taxon, 64: 703-713. DOI:10.12705/644.3 |
Govaerts, R., Friesen, N., Fritsch, R., Snijman, D. A., Marcucci, R., SilverstoneSopkin, P. A., Brullo, S., 2016. World Checklist of Allium. Royal Botanic Gardens. http://apps.kew.org/wcsp/.
|
Graham C.H., Carnaval A.C., Cadena C.D., Zamudio K.R., Roberts T.E., Parra J.L., McCain C.M., Bowie R.C., Moritz C., Baines S.B., 2014. The origin and maintenance of montane diversity:integrating evolutionary and ecological processes. Ecography, 37: 711-719. DOI:10.1111/ecog.2014.v37.i8 |
Gregory, M., Fritsch, R. M., Friesen, N. W., Khassanov, F. O., McNeal, D. W., 1998. Nomenclator alliorum: Allium names and synonyms: a world guide. Royal Botanic Gardens, Kew, ISBN 1900347644, p. 83.
|
Haffer J., 1969. Speciation in Amazonian forest birds. Science, 165: 131-137. DOI:10.1126/science.165.3889.131 |
Heled J., Bouckaert R.R., 2013. Looking for trees in the forest:summary tree from posterior samples. BMC Evol. Biol., 13: 1. DOI:10.1186/1471-2148-13-1 |
Herden T., Hanelt P., Friesen N., 2016. Phylogeny of Allium Lsubgenus Anguinum (G.Don. ex W.D.J. Koch) N. Friesen (Amaryllidaceae). Mol. Phylogenet. Evol., 95: 79-93. DOI:10.1016/j.ympev.2015.11.004 |
Hoorn C., Mosbrugger V., Mulch A., Antonelli A., 2013. Biodiversity from mountain building. Nat. Geosci., 6: 154. DOI:10.1038/ngeo1742 |
Huang D.-Q., Yang J.-T., Zhou C.-J., Zhou S.-D., He X.-J., 2014. Phylogenetic reappraisal of Allium subgenus Cyathophora (Amaryllidaceae) and related taxa, with a proposal of two new sections. J. Plant Res., 127: 275-286. DOI:10.1007/s10265-013-0617-8 |
Hughes C.E., Atchison G.W., 2015. The ubiquity of alpine plant radiations:from the Andes to the Hengduan Mountains. New Phytol., 207: 275-282. DOI:10.1111/nph.2015.207.issue-2 |
Katoh K., Standley D.M., 2013. MAFFT multiple sequence alignment software version 7:improvements in performance and usability. Mol. Biol. Evol., 30: 772-780. DOI:10.1093/molbev/mst010 |
Kaufman D.S., Manley W.F., 2004. Pleistocene maximum and Late Wisconsinan glacier extents across Alaska, USA. Dev. Quat. Sci., 2: 9-27. |
Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., 2012. Geneious basic:an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28: 1647-1649. DOI:10.1093/bioinformatics/bts199 |
Lagomarsino L.P., Condamine F.L., Antonelli A., Mulch A., Davis C.C., 2016. The abiotic and biotic drivers of rapid diversification in Andean bellflowers (Campanulaceae). New Phytol., 210: 1430-1442. DOI:10.1111/nph.13920 |
Li M.J., Tan J.B., Xie D.F., Huang D.Q., Gao Y.D., He X.J., 2016. Revisiting the evolutionary events in Allium subgenus Cyathophora (Amaryllidaceae):insights into the effect of the Hengduan Mountains Region (HMR) uplift and Quaternary climatic fluctuations to the environmental changes in the Qinghai-Tibet Plateau. Mol. Phylogenet. Evol., 94: 802-813. DOI:10.1016/j.ympev.2015.10.002 |
Li Q.-Q., Zhou S.-D., He X.-J., Yu Y., Zhang Y.-C., Wei X.-Q., 2010. Phylogeny and biogeography of Allium (Amaryllidaceae:Allieae) based on nuclear ribosomal internal transcribed spacer and chloroplast rps16 sequences, focusing on the inclusion of species endemic to China. Ann. Bot., 106: 709-733. DOI:10.1093/aob/mcq177 |
Linder H.P., 2008. Plant species radiations:where, when, why? Philos. Trans. R. Soc.B Biol. Sci., 363: 3097-3105. DOI:10.1098/rstb.2008.0075 |
Mabberley D.J., 2008. Mabberley's Plant-Book:a Portable Dictionary of Plants, Their Classifications and Uses. Camb. Univ. Press.
|
Maddison W.P., Midford P.E., Otto S.P., 2007. Estimating a binary character's effect on speciation and extinction. Syst. Biol., 56: 701-710. DOI:10.1080/10635150701607033 |
Magallón S., Gomez-Acevedo S., Sanchez-Reyes L.L., Hernandez-Hernandez T., 2015. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol., 207: 437-453. DOI:10.1111/nph.2015.207.issue-2 |
Manafzadeh S., Staedler Y.M., Conti E., 2016. Visions of the past and dreams of the future in the Orient:the Irano-Turanian region from classical botany to evolutionary studies. Biol. Rev.. DOI:10.1111/brv.12287 |
Matzke N.J., 2014. Model selection in historical biogeography reveals that founderevent speciation is a crucial process in Island clades. Syst. Biol., 63: 951-970. DOI:10.1093/sysbio/syu056 |
Moore B.R., Höhna S., May M.R., Rannala B., Huelsenbeck J.P., 2016. Critically evaluating the theory and performance of Bayesian analysis of macroevolutionary mixtures. Proc. Natl. Acad. Sci. U. S. A., 113: 9569-9574. DOI:10.1073/pnas.1518659113 |
Mosbrugger, V., Favre, A., Muellner-Riehl, A. N., Päckert, M., Mulch, A., 2018. Ceno-zoic evolution of geo-biodiversity in the Tibeto-Himalayan region. In: Hoorn, C., Antonelli, A. (Eds. ), Mountains, Climate, and Biodiversity. Wiley-Blackwell, ISBN 978-1-119-15987-2 (in press).
|
Nee S., May R.M., Harvey P.H., 1994. The reconstructed evolutionary process. Phil.Trans. R. Soc. London B, 344: 305-311. DOI:10.1098/rstb.1994.0068 |
Palazzesi L., Barreda V.D., Cuitiño J.I., Guler M.V., Tellería M.C., Ventura Santos R., 2014. Fossil pollen records indicate that Patagonian desertification was not solely a consequence of Andean uplift. Nat. Commun., 5: 3558. |
Pattengale N.D., Alipour M., Bininda-Emonds O.R., Moret B.M., Stamatakis A., 2010. How many bootstrap replicates are necessary? J. Comput. Biol., 17: 337-354. DOI:10.1089/cmb.2009.0179 |
R. Core Team, 2015. R: a language and environment for statistical computing. Vienna, Austria.
|
Rabosky D.L., 2014. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS ONE, 9: e89543. DOI:10.1371/journal.pone.0089543 |
Rabosky D.L., Grundler M., Anderson C., Shi J.J., Brown J.W., Huang H., Larson J.G., 2014. BAMMtools:an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol., 5: 701-707. DOI:10.1111/mee3.2014.5.issue-7 |
Rabosky D.L., Mitchell J.S., Chang J., 2017. Is BAMM flawed? Theoretical and practical concerns in the analysis of multi-rate diversification models. Syst. Biol.. DOI:10.1093/sysbio/syx037 |
Ree R.H., 2005. Detecting the historical signature of key innovations using stochastic models of character evolution and cladogenesis. Evolution, 59: 257-265. DOI:10.1111/evo.2005.59.issue-2 |
Ree R.H., Smith S.A., 2008. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst. Biol., 57: 4-14. DOI:10.1080/10635150701883881 |
Renner S.S., 2016. Available data point to a 4-km-high Tibetan Plateau by 40 Ma, but 100 molecular-clock papers have linked supposed recent uplift to young node ages. J. Biogeogr., 43: 1479-1487. DOI:10.1111/jbi.12755 |
Ronquist F., Cannatella D., 1997. Dispersal-vicariance analysis:a new approach to the quantification of historical biogeography. Syst. Biol., 46: 195-203. DOI:10.1093/sysbio/46.1.195 |
Ronquist F., Teslenko M., van der Mark P., Ayres D.L., Darling A., Höhna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J.P., 2012. MrBayes 3.2:efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol., 61: 539-542. DOI:10.1093/sysbio/sys029 |
Schwery O., Onstein R.E., Bouchenak-Khelladi Y., Xing Y., Carter R.J., Linder H.P., 2015. As old as the mountains:the radiations of the Ericaceae. New Phytol., 207: 355-367. DOI:10.1111/nph.2015.207.issue-2 |
Sennikov A.N., Seregin A.P., 2015. What happens to Allium saxatile M. Bieb.(Amaryllidaceae)? An unknown story of the well-known name. Taxon, 64: 1294-1300. DOI:10.12705/646.11 |
Smith, S. Y., 2013. The fossil record of non-commelinid monocots. In: Wilkin, P., Mayo, S. (Eds. ), Early Events in Monocot Evolution, Systematics Association Special Volume Series. Cambridge University Press, Cambridge. doi: 10.1017/CBO9781139002950.003.
|
Silvestro D., Schnitzler J., Zizka G., 2011. A Bayesian framework to estimate diversification rates and their variation through time and space. BMC Evol.Biol., 11. |
Silvestro, D., Schnitzler, J., 2018. Inferring macroevolutionary dynamics in mountain systems from fossils. In: Hoorn, C., Antonelli, A. (Eds. ), Mountains, Climate, and Biodiversity. Wiley-Blackwell, ISBN 978-1-119-15987-2.
|
Stamatakis A., 2014. RAxML version 8:a tool for phylogenetic analysis and postanalysis of large phylogenies. Bioinformatics, 30: 1312-1313. DOI:10.1093/bioinformatics/btu033 |
Tang Z., Wang Z., Zheng C., Fang J., 2006. Biodiversity in China's mountains. Front.Ecol. Environ., 4: 347-352. DOI:10.1890/1540-9295(2006)004[0347:BICM]2.0.CO;2 |
Wang C., Dai J., Zhao X., Li Y., Graham S.A., He D., Ran B., Meng J., 2014. Outwardgrowth of the Tibetan Plateau during the Cenozoic:a review. Tectonophysics, 621: 1-43. DOI:10.1016/j.tecto.2014.01.036 |
Xing Y., Ree R.H., 2017. Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proc. Natl. Acad. Sci. U. S. A., 114: E3444-E3451. DOI:10.1073/pnas.1616063114 |
Xu, J., Kamelin, R., 2000. In: Wu, Z., Raven, P. H., Deyuan, H. (Eds. ), Allium L. Flora of China, vol. 24. Missouri Botanical Garden Press & Science Press, St. Louis, Beijing, pp. 165-202.
|
Xu B., Li Z., Sun H., 2014. Plant diversity and floristic characters of the alpine subnival belt flora in the Hengduan Mountains, SW China. J. Syst. Evol., 52: 271-279. DOI:10.1111/jse.v52.3 |
Zhang D.C., Ye J.X., Sun H., 2016. Quantitative approaches to identify floristic units and centres of species endemism in the Qinghai-Tibetan Plateau, south-western China. J. Biogeogr., 43: 2465-2476. DOI:10.1111/jbi.12819 |