



2. 吉林大学水资源与环境研究所, 长春 130021
2. Institute of Water Resources and Environment, Jilin University, Changchun 130021, China
0 引言
有机氯代烃(volatile chlorinated hydrocarbon,HVOC,如三氯乙烯、1, 1, 1-三氯乙烷)作为干洗剂、金属清洗剂、萃取剂和化工原料等广泛应用于工业和商业中。然而,有机氯代烃的大量生产、使用、储存以及处置不当,使其进入土壤、地下水,成为地下环境中常见的有机污染物[1]。由于有机氯代烃在土壤介质中迁移时可移动性差、难自然衰减,且本身具有一定的毒性和致癌性,因此对人类健康和生态环境造成了严重的危害[2]。
原位化学氧化技术(in situ chemical oxidation,ISCO)被认为是去除土壤及地下水中有机污染物切实可行、经济高效的修复方法之一[3]。近年来,过硫酸盐(S2O82-,persulfate,PS)作为一种新型的氧化剂,学者们对其进行了深入的研究[4]。过硫酸盐在水中电离产生SO4-·,其标准氧化还原电位E0=2.60 V[5-6],接近羟基自由基(OH·,E0=2.70 V)[7-8]。活化方式主要有热活化[9-10]、过渡金属离子活化[11]、光活化及其他复合活化方式[12-14]。其中,热活化过硫酸盐技术虽然能耗较高,但其活化过硫酸盐效果最好,也是一种清洁型污染物处理技术[15]。热活化过硫酸盐活化机理为S2O82-+heat→2SO4-·。Huang等[15-16]利用热活化方式对过硫酸钠进行了活化,发现活化后过硫酸钠可高级氧化降解59种挥发性有机污染物。随后,Liang等[17-18]研究发现活化过硫酸钠氧化降解TCE的效果随着温度的升高而增加。此外,加热过程还可以改变土壤对有机物的吸附量[19],加速土壤中(尤其是黏土类土壤)氯代烃的解吸过程,提高氯代烃和过硫酸盐的有效接触,进而提高氯代烃的去除效果。然而,由于土壤介质中含有矿物成分、有机质或无机离子等组分,这些组分会与过硫酸盐发生反应,导致过硫酸盐的部分自我消耗。因此,土壤中挥发性氯代烃的氧化降解过程往往需要投加过多的过硫酸盐[20]。此外,土壤类型不同,其组分含量不同,对过硫酸盐的自我消耗影响也不同。由此可见,掌握不同土壤类型中过硫酸盐的分解反应速率对原位化学氧化技术体系的成功设计至关重要。然而,目前多数研究仅针对单一土壤介质中有机污染物的氧化降解效果,而针对不同土壤类型(尤其是黏土类土壤)中挥发性氯代烃的氧化降解效果及土壤组分对过硫酸盐的自我分解反应速率的影响研究鲜有报道。
本文拟开展热活化过硫酸盐氧化降解两种典型土壤(砂类土壤、黏土类土壤)中挥发性氯代烃(三氯乙烯(TCE)、三氯乙烷(TCA)、顺式-1, 2 -二氯乙烯(cis-1, 2-DCE)、1, 2-二氯乙烷(1, 2-DCA)的研究,探究过硫酸盐与氯代烃和土壤介质之间相互作用过程中的自身分解反应情况,进一步解析不同土壤介质中有机质和无机组分对过硫酸盐自身消耗和氯代烃氧化降解效果的影响作用。
1 材料与方法 1.1 设备与试剂实验所用设备为ZWY-240恒温振荡器、离子色谱仪(ICS-2100,DIONEX USA)、7890A气相色谱仪(Agilent USA)。
实验所用试剂有三氯乙烯、三氯乙烷、顺式-1, 2 -二氯乙烯、1, 2-二氯乙烷(纯度≥99%)和过硫酸钠(分析纯),实验用水均为超纯水。
本实验采用的砂类土壤样品取自于长春地表土壤,而黏土类土壤则取自于较深层土壤。2类土壤的粒径分布如图 1所示。由图 1可知,砂类土壤的粒径主要分布在150 ~400 μm之间,黏土类土壤的粒径主要分布在5~100 μm之间。黏土土壤具有较大的比表面积,因而吸附能力应强于砂类土土壤。

|
| 图 1 土壤粒径分布图 Figure 1 Particle size distribution of sandy soil and clayey soil |
|
|
本实验采用恒温摇瓶实验,实验所用的反应瓶为30 mL棕色瓶,瓶盖内衬聚四氟乙烯隔垫。首先按照1, 2-DCA、TCA、cis-1, 2-DCE、TCE比例为1:1:1:1配制氯代烃母液(质量浓度为1 011.9 mg/L),母液中1, 2-DCA、TCA、cis-1, 2-DCE、TCE的质量浓度分别为0.31、0.30、0.32、0.37 g/L(其溶解度分别为8.7、1.4、3.5、1.0 g/L),并配制过硫酸钠母液10 g/L。
热活化过硫酸盐降解氯代烃实验共分2组,一组为研究土壤中过硫酸盐氧化降解氯代烃的实验(实验Ⅰ),另一组为研究相同条件下土壤中过硫酸盐的自我消耗实验(实验Ⅱ)。两组采用土壤样品均分为黏土类土壤和砂类土壤。实验Ⅰ中,首先向每个30 mL棕色瓶中加入20 g土壤样品,压盖密封;再加入90 μL氯代烃母液与少量超纯水,使土壤与污染物混合均匀;然后再向各瓶中注入9 mL过硫酸钠母液,加超纯水至满摇匀,使得反应瓶内最终过硫酸盐质量浓度为30 g/L,氯代烃质量浓度为4.17 g/L。实验Ⅱ中,以超纯水代替9 mL的过硫酸钠溶液,其余步骤相同。每组实验不同条件反应瓶各设置3组平行样品,反应瓶在50 ℃下恒温震荡11 d,每天取样检测。
同时,为探究土壤中过硫酸盐的自我消耗,设立了过硫酸盐与土壤之间的反应实验,实验反应瓶依旧采用30 mL棕色瓶,瓶盖内衬聚四氟乙烯隔垫。向每个30 mL棕色瓶中加入20 g土壤样品(黏土类土壤、砂类土壤),各注入9 mL过硫酸钠母液,使得反应瓶内最终过硫酸盐浓度为30 g/L,加超纯水至满压盖密封摇匀。设置3组平行样品,反应瓶在50 ℃摇床中恒温振荡。
1.3 测试方法过硫酸钠的测定采用UV-碘量法[21]。
以最终每个反应瓶解吸出的氯代烃质量浓度为依据考察氯代烃的降解效果,挥发性氯代烃的测定采用吹扫捕集-气相色谱法。分析条件为:Agilent 7693自动进样器,色谱柱为HP-5毛细管柱(长30 m,内径0.32 mm,膜厚0.25 μm),进样口及ECD(电子捕获检测器)温度分别为150 ℃、250 ℃,柱温恒温40 ℃,柱流量2 mL/min,分流比10:1。
2 结果与讨论 2.1 不同土壤类型介质中氯代烃的降解效果实验Ⅰ和Ⅱ效果如图 2所示。在4种有机氯代烃中,cis-1, 2-DCE与TCE属于氯代烯烃,其双键在自由基攻击下易断裂而分解;1, 2-DCA与TCA属于氯代烷烃,化学性质较为稳定且不易降解,且1, 2-DCA为合成TCA的中间体,所以TCA较1, 2-DCA更加稳定。由图 2可以看出,1, 2-DCA、cis-1, 2-DCE、TCA、TCE均得到了不同程度的降解。50 ℃砂类土壤中4种氯代烃降解效果依次为25%、89%、5%、61%;黏土类土壤中四种氯代烃降解效果依次为35%、86%、8%、63%。同种土壤介质中的4种氯代烃降解效果差异较大,其降解效率呈现cis-1, 2-DCE>TCE>1, 2-DCA>TCA的规律,与氯代烃的氧化降解研究结果相符合[22-23]。具有亲电性的硫酸根自由基通过电子的转移、夺氢或加氢来攻击有机污染物分子。随着有机氯代烃分子中C上的Cl逐渐被取代,C-H的强度随之增强,氯代烃的稳定性也会增强。由于烯烃(TCE、cis-1, 2-DCE)是不饱和烃,C=C键中后一个键易断裂,能够分别与其他原子或原子团结合成新的共价键,因而TCE、cis-1, 2-DCE具有良好的降解效果;而烷烃(1, 2-DCA、TCA)单键为饱和状态,性质稳定,因而降解效果较差。同时,观察到1, 2-DCA的降解效果要明显优于TCA,这是由于TCA分子结构最为稳定,且易吸附于土壤中,降解效果要低于其他3种氯代烃污染物。由图 2可以看出,cis-1, 2-DCE、TCE的降解效率较高,可达60%以上,但TCA、1, 2-DCA的降解效率却低于50%,总体来说降解效果并不理想。反应瓶溶液中Cl-平均质量浓度的变化规律如图 3所示。

|
| 图 2 砂土类土壤和黏土类土壤中四种氯代烃的降解效率 Figure 2 Degradation rate of chlorinated hydrocarbons with sandy soil and clayey soil |
|
|

|
| 图 3 过硫酸盐氧化降解氯代烃反应中土壤中溶液氯离子质量浓度变化规律 Figure 3 Free chloride concentration in the oxidative degradation of chlorinated hydrocarbons by PS of sandy soil and clayey soil |
|
|
由图 3可以看到,反应过程中溶液中Cl-的生成量呈上升趋势,开始的7 d内呈快速增长趋势,7 d之后Cl-的增长趋于稳定。Cl-的增长趋势与4种氯代烃的降解过程对应相符。砂类土壤和黏土类土壤溶液中的Cl-最终质量浓度分别为17.3 mg/L和18.1 mg/L,对应最终的氯代烃降解效果。
分别对土壤中过硫酸盐的自我消耗研究各个反应瓶中反应后的溶液进行离子色谱检测。结果表明砂类、黏土类两种土壤均未检出Cl-,说明热活化过硫酸盐降解氯代烃实验中产生的Cl-均来自氯代烃的降解。
综合图 2、3可知,溶液中的氯代烃经过硫酸盐氧化产生Cl-,黏土类土壤介质中产生的氯离子量高于砂类土壤中产生的氯离子量,同时说明了黏土类的降解效果。黏土类土壤相较于砂土类土壤中氯代烃的降解效果要更优,因为黏土类土壤粒径小、比表面积大、吸附能力强,这种土壤性质的差异有利于反应的进行。
2.2 土壤介质类型和组分对过硫酸盐自身消耗的影响 2.2.1 砂类、黏土类土壤介质中过硫酸盐的自身消耗图 4为砂类、黏土类土壤中过硫酸盐氧化降解氯代烃和过硫酸盐的自我消耗反应过程中溶液过硫酸盐质量浓度变化。图 4a中,溶液中过硫酸盐的质量浓度随着时间的推移逐步降低,最终趋于平缓,说明分解速率由快变慢。砂类土壤和黏土类土壤中最终的过硫酸盐质量浓度分别为5.37、7.40 g/L,过硫酸盐的消耗率分别为82.1%和75.3% (表 1),砂类土壤中过硫酸盐的消耗量要高于黏土类土壤。图 4b中,砂类土壤和黏土类土壤中最终的过硫酸盐质量浓度分别为5.56 g/L、9.68 g/L,过硫酸盐的消耗率为81.3%和66.7%(表 1)。砂类和黏土类土壤中在没有污染物的条件下过硫酸盐依旧有大量的自我消耗,过硫酸盐的自我消耗主要来源于热活化过程中过硫酸盐在外加热能的条件下直接转化为硫酸根自由基[14]。同时,砂土类土壤自身过硫酸盐消耗要高于黏土类土壤,这是由于砂土类土壤颗粒之间的空隙较大,过硫酸盐在颗粒间自由流动会使得颗粒表面的矿物质容易与过硫酸盐充分接触;而黏土类土壤颗粒间空隙较小,过硫酸盐不容易渗透入黏土结构中,只能与表层的黏土接触,从而过硫酸盐消耗较低。

|
| 图 4 过硫酸盐氧化降解氯代烃反应(a)和过硫酸盐的自我消耗反应(b)土壤中溶液过硫酸盐质量浓度变化 Figure 4 PS variation in the oxidative degradation(a)and in the self depletion (b)of chlorinated hydrocarbons by PS in sandy soil and clayey soil |
|
|
| 研究批次 | 土壤介质类型 | 过硫酸盐初始质量浓度/(g/L) | 过硫酸盐最终质量浓度/(g/L) | 过硫酸盐消耗率/% |
| 过硫酸盐+土壤+氯代烃 | 砂类土壤 | 30.00 | 5.37 | 82.1 |
| 黏土类土壤 | 30.00 | 7.40 | 75.3 | |
| 过硫酸盐+土壤 | 砂类土壤 | 30.00 | 5.56 | 81.3 |
| 黏土类土壤 | 30.00 | 9.68 | 66.7 |
通过含氯代烃(图 4a)和不含氯代烃(图 4b)的土壤-过硫酸盐系统过硫酸盐消耗水平差异可以看出,当体系中没有氯代烃时,过硫酸盐在50 ℃下主要与土壤介质组分发生反应并被充分消耗;而当体系中加氯代烃时,氯代烃与土壤介质组分竞争过硫酸盐,过硫酸盐会同时与二者发生反应。从图 4中可以看出氯代烃的加入对两种体系下的过硫酸盐消耗量影响不大,说明氯代烃并不是体系中过硫酸盐的唯一消耗源,氯代烃会与土壤介质组分两者竞争消耗过硫酸盐。这种竞争关系的存在说明除了氯代烃,其他因素如土壤介质中金属离子、土壤有机质等对过硫酸盐消耗产生的影响不可忽视[24-25]。
由表 1可知,两组研究中砂类土壤、黏土类土壤所对应的消耗率差值分别为0.8%和8.6%。对比土壤、过硫酸盐对氯代烃的降解效果研究中的氯代烃降解率可以进一步判断,土壤介质中可以与过硫酸盐反应的组分与氯代烃在与过硫酸盐的反应中应存在竞争关系,且土壤介质组分消耗过硫酸盐占据主导地位,因而造成氯代烃的降解效果并不理想。
定义OE(oxidation efficiency)为单位过硫酸盐氧化降解有机氯代烃的量,其计算公式如下:
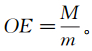 (1)
(1) 式中:M为该反应体系中消耗的有机氯代烃总物质的量(mmol);m为该反应体系消耗的过硫酸盐的总质量(g)。通过计算可以得出OE砂类土壤 = 0.34 mmol/g,OE黏土类土壤=0.41 mmol/g,OE黏土类土壤>OE砂类土壤。可以看出由于土壤类型不同,OE不同,即黏土类土壤中过硫酸盐氧化降解有机氯代烃的效率要高于砂类土壤。其结果与土壤的理化性质及其组分与过硫酸盐和有机氯代烃相互之间的影响有关。
2.2.2 土壤介质性质及组分对过硫酸盐消耗的影响研究所用砂类土壤、黏土类土壤(反应前测定)比表面积和粒度分布特征如表 2所示,经过X射线荧光光谱分析,获得反应前后砂类和黏土类土壤样本中的各项组分质量分数,如表 3、表 4所示。
| 土壤颗粒性质 | 比表面积/(m2/g) | 中值粒径/μm | φB/ % | |||
| 砾石 | 粉砂+砂 | 细粉砂 | 黏土 | |||
| 黏土类土壤 | 0.24 | 25.1 | 0 | 56 | 37 | 7 |
| 砂类土壤 | 0.07 | 241.3 | 0 | 90 | 8 | 2 |
| 注:不同颗粒类型粒径d范围分别定义为:砾石.d>2.000 mm;粉砂+砂.d为0.020~2.000 mm;细粉砂.d为0.002~0.020 mm;黏土d < 0.002 mm。 | ||||||
| 土壤类型 | 测定时间 | wB/% | ||
| 总有机碳 | 铁元素 | 锰元素 | ||
| 黏土类土壤 | 反应前 | 0.47 | 5.13 | 0.20 |
| 反应后 | 0.13 | 3.15 | 0.09 | |
| 砂类土壤 | 反应前 | 0.29 | 2.22 | 0.08 |
| 反应后 | 0.05 | 0.43 | 0.00 | |
| 土壤类型 | 有机质 | 铁元素 | 锰元素 | 1, 2-DCA | cis-1, 2-DCE | TCA | TCE | 过硫酸盐 | 过硫酸盐(过硫酸盐自我消耗反应) |
| 砂类土壤 | 81.3 | 80.5 | 100.0 | 25.0 | 89.0 | 5.0 | 61.0 | 82.1 | 81.3 |
| 黏土类土壤 | 72.6 | 38.6 | 55.0 | 35.0 | 86.0 | 8.0 | 63.0 | 75.3 | 66.7 |
由表 2可以看出:砂类土壤的中值粒径为241.3 μm,而黏土类土壤的中值粒径为25.1 μm,黏土类土壤粒径远远小于砂类粒径;黏土类土壤和砂类土壤的比表面积分别为0.24和0.07 m2/g,黏土类土壤的比表面积高于砂类土壤,因此具备很强的吸附能力。氯代烃在土壤和溶液之间存在一个吸附解吸的动态平衡[26-27]。根据土壤中过硫酸盐氧化降解氯代烃反应结果可知,氯代烃并没有被充分氧化降解。结合两种土壤介质各项参数说明部分氯代烃都被土壤吸附,从土壤中解吸出来的氯代烃以液相的形式存在,在氧化降解过程中溶液中的氯代烃和土壤中吸附的氯代烃的迁移转化保持动态平衡。
由表 3可以看出:反应前黏土类土壤中含有铁元素5.13%、锰元素0.20%,而砂类土壤则含有铁元素2.22%、锰元素0.08%,黏土类土壤中这两项金属元素质量分数高于砂类土壤;黏土类土壤和砂类土壤中总有机碳的质量分数分别为0.47%和0.29%,前者总有机碳质量分数较高。
反应后:黏土类和砂类两种土壤介质中的总有机碳分别为0.13%和0.05%,较反应前明显减少,说明土壤中的有机质也参与了过硫酸盐的反应;黏土类和砂类土壤介质中铁元素、锰元素的质量分数分别为3.15%和0.43%,0.09%与0.00%,反应后明显降低。由实验所得数据可以看出,铁元素、锰元素是具有活化过硫酸盐能力的过渡金属元素,在热活化过硫酸盐降解氯代烃的过程中参与反应并消耗了过硫酸盐[28-30],其作用原理如下(Me为过渡金属元素)[31]:
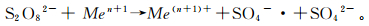
因此,土壤中有机质与铁、锰两种过渡金属元素的变化均可以说明其作为土壤中重要组分参与了活化反应。
参与土壤中过硫酸盐氧化降解氯代烃反应的土壤中组分消耗率、氯代烃降解效率以及过硫酸盐消耗率如表 4所示。
由表 4可以看出,在过硫酸盐氧化降解氯代烃反应中,砂类土壤中各组分的消耗率均高于黏土类土壤中相应组分的消耗率。其中:有机质在反应前后的消耗率为81.3%和72.6%;铁元素、锰元素在反应前后的消耗率分别为80.5%和38.6%与100.0%和55.0%。而这些土壤组分不会直接与氯代烃发生反应,其减少说明这些土壤组分均参与到过硫酸盐的自我消耗反应中;砂类土壤中各组分的消耗量均高于黏土类土壤中相应组分的消耗量,且砂类土壤中过硫酸盐的自我消耗率高于黏土类土壤,说明砂类土壤中过硫酸盐的自我消耗强于黏土类土壤。
结合对OE值的对比研究、参与反应各物质反应前后的质量分数对比、土壤中过硫酸盐氧化降解氯代烃反应和过硫酸盐自我消耗反应研究及各物质消耗率可以看出,不同土壤条件下过硫酸盐的消耗量及有机氯代烃的降解效果均体现出了不同;同时,对比反应前后土壤组分的变化也可以看出土壤介质组分也参与了过硫酸盐的消耗。因此土壤中各个组分及4种有机氯代烃在与过硫酸盐的反应中存在竞争关系。
综上所述,在不同类型土壤介质环境中热活化过硫酸盐降解氯代烃体系中,土壤介质组分对过硫酸盐的消耗、氯代烃的降解效果具有重要的影响。因此,针对实际有机氯代烃污染场地,采用过硫酸盐氧化技术进行修复时,过硫酸盐的实际投加量要远高于化学计量值,需充分考虑到土壤组分对过硫酸盐自身的消耗作用。
3 结论1) 在50 ℃条件下,热活化过硫酸盐可以有效降解土壤中有机氯代烃,在砂类土壤和黏土类土壤中1, 2-DCA、cis-1, 2-DCE、TCA、TCE降解效果依次为25%、89%、5%、61%,和35%、86%、8%、63%。黏土类土壤中氯代烃的降解效果优于砂类土壤中,且4种氯代烃的降解顺序为cis-1, 2-DCE>TCE>1, 2-DCA>TCA。
2) 在热活化过硫酸盐降解氯代烃的反应中,土壤中的有机质、铁元素、锰元素等物质均参与反应并消耗过硫酸盐。土壤中过硫酸盐氧化降解氯代烃反应研究中,黏土和细砂介质中过硫酸盐消耗分别为75.3%和82.1%;土壤中过硫酸盐的自我消耗反应研究中,土壤组分会大量消耗体系中的过硫酸盐,黏土和细砂2种土壤中消耗过硫酸盐分别为67.7%和81.3%,且土壤组分(有机质、铁元素、锰元素等)与氯代烃之间在消耗过硫酸盐反应上存在竞争关系,因而造成4种氯代烃的降解效果并不理想。因此,土壤介质组分对过硫酸盐的利用效率和有机氯代烃的降解具有重要的影响。
| [1] | Pankow J F, Cherry J A. Dense Chlorinated Solvents and Other DNAPLs in Groundwater: History, Behavior, and Remediation. [M]. Waterloo Press: Portland, OR, 1996: 522. |
| [2] | Bianchi A P, Varney M S, Phillips J. Analysis of Volatile Organic Compounds in Estuarine Sediments Using Dynamic Headspace and Gas Chromatography:Mass Spectrometry[J]. Journal of Chromatography:A, 1991, 542(1): 413-450. |
| [3] |
梁花梅, 孙德栋, 郑欢, 等. 利用硫酸根自由基处理剩余污泥[J].
大连工业大学学报, 2013, 32(1): 47-50.
Liang Huamei, Sun Dedong, Zheng Huan, et al. Treatment of Excess Sludge with Sulfate Free Radical[J]. Journal of Dalian Polytechnic University, 2013, 32(1): 47-50. |
| [4] |
刘红梅, 褚华强, 陈家斌, 等. 过硫酸盐在地下水和土壤修复中的应用[J].
现代化工, 2015, 35(4): 42-46.
Liu Hongmei, Chu Huaqiang, Chen Jiabin, et al. Application of Persulfate in Remediating Groundwater and Soil[J]. Modern Chemical Industry, 2015, 35(4): 42-46. |
| [5] | Huie R E, Clifton C L, Neta P. Electron Transfer Reaction Rates and Equilibria of the Carbonate and Sulfate Radical Anions[J]. International Journal of Radiation Applications & Instrumentation:Part C:Radiation Physics & Chemistry, 1991, 38(5): 477-481. |
| [6] | House D A. Kinetics and Mechanism of Oxidations by Peroxydisulfate[J]. Chemical Reviews, 1962, 62(3): 185-203. DOI:10.1021/cr60217a001 |
| [7] |
龙安华, 雷洋, 张晖. 活化过硫酸盐原位化学氧化修复有机污染土壤和地下水[J].
化学进展, 2014, 26(5): 898-908.
Long Anhua, Lei Yang, Zhang Hui. In Situ Chemical Oxidation of Organic Contaminated Soil and Groundwater Using Activated Persulfate Process[J]. Progress in Chemistry, 2014, 26(5): 898-908. |
| [8] | Buxton G V, Greenstock C L, Helman W P, et al. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (·OH/·O-) in Aqueous Solution[J]. Journal of Physical & Chemical Reference Data, 1988, 17(2): 513-886. |
| [9] | Mora V C, Rosso J A, M Rtire D O, et al. Phenol Depletion by Thermally Activated Peroxydisulfate at 70℃[J]. Chemosphere, 2011, 84(9): 1270-1275. DOI:10.1016/j.chemosphere.2011.04.062 |
| [10] | Waldemer R H, Tratnyek P G, Johnson R L, et al. Oxidation of Chlorinated Ethenes by Heat-Activated Persulfate:Kinetics and Products[J]. Environmental Science & Technology, 2007, 41(3): 1010-1015. |
| [11] | Liu C S, Shih K, Sun C X, et al. Oxidative Degradation of Propachlor by Ferrous and Copper Ion Activated Persulfate[J]. Science of the Total Environment, 2012, 416(2): 507-512. |
| [12] |
杨世迎, 杨鑫, 王萍, 等. 过硫酸盐高级氧化技术的活化方法研究进展[J].
现代化工, 2009, 29(4): 13-19.
Yang Shiying, Yang Xin, Wang Ping, et al. Advances in Persulfate Oxidation Activation Methods of Persulfate Oxidation[J]. Modern Chemical Industry, 2009, 29(4): 13-19. |
| [13] | Yang Shiying, Wang Ping, Yang Xin, et al. A Novel Advanced Oxidation Process to Degrade Organic Pollutants in Wastewater:Microwave-Activated Persulfate Oxidation[J]. J Environment Science, 2009, 21(9): 1175-1180. DOI:10.1016/S1001-0742(08)62399-2 |
| [14] | Gao Yuqiong, Gao Naiyun, Deng Yang, et al. Ultraviolet (UV) Light-Activated Persulfate Oxidation of Sulfamethazine in Water[J]. Chem Eng J, 2012, 195/196: 248-253. DOI:10.1016/j.cej.2012.04.084 |
| [15] | Mora V C, Rosso J A, Galo C L R, et al. Thermally Activated Peroxydisulfate in the Presence of Additives:A Clean Method for the Degradation of Pollutants[J]. Chemosphere, 2009, 75(10): 1405-1409. DOI:10.1016/j.chemosphere.2009.02.038 |
| [16] | Huang K C, Zhao Z, Hoag G E, et al. Degradation of Volatile Organic Compounds with Thermally Activated Persulfate Oxidation[J]. Chemosphere, 2005, 61(4): 551-560. DOI:10.1016/j.chemosphere.2005.02.032 |
| [17] | Liang C, Bruell C J, Marley M C, et al. Persulfate Oxidation for In Situ Remediation of TCE:Ⅰ:Activated by Ferrous Ion with and Without a Persulfate:Thiosulfate Redox Couple[J]. Chemosphere, 2004, 55(9): 1213-1223. DOI:10.1016/j.chemosphere.2004.01.029 |
| [18] | Liang C, Bruell C J, Marley M C, et al. Persulfate Oxidation for in Situ Remediation of TCE:Ⅱ:Activated by Chelated Ferrous Ion[J]. Chemosphere, 2004, 55(9): 1225-1233. DOI:10.1016/j.chemosphere.2004.01.030 |
| [19] | Vance G F, David M B. Dissolved Organic Carbon and Sulfate Sorption by Spodosol Mineral Horizons[J]. Soil Science, 1992, 154(2): 136-144. DOI:10.1097/00010694-199208000-00007 |
| [20] | Bougie S, Dub J. Oxydation des Isomères de Iichlo-robenzène à L'aide du Persulfate de Sodium Soumis à Une Activation Thermique[J]. Journal of Environmental Engineering & Science, 2007, 6(4): 397-407. |
| [21] | Liang Chenju, Huang Chiufen, Mohanty N, et al. A Rapid Spectrophotometric Determination of Persulfate Anion in ISCO[J]. Chemosphere, 2008, 73(9): 1540-1543. DOI:10.1016/j.chemosphere.2008.08.043 |
| [22] | Johnson R L, Tratnyek P G, Johnson R O. Per-sulfate Persistence under Thermal Activation Conditions[J]. Environmental Science & Technology, 2008, 42(24): 9350-9356. |
| [23] | Wu Xiaoliang, Gu Xiaogang, Lu Shuguang, et al. Strong Enhancement of Trichloroethylene Degradation in Ferrous Ion Activated Persulfate System by Promoting Ferric and Ferrous Ion Cycles with Hydroxylamine[J]. Separation & Purification Technology, 2015, 147: 186-193. |
| [24] | Anipsitakis G P, Dionysiou D D. Radical Generation by the Interaction of Transition Metals with Common Oxidants[J]. Environmental Science & Technology, 2004, 38(13): 3705-3712. |
| [25] | Ko S, Crimi M, Marvin B K, et al. Comparative Study on Oxidative Treatments of NAPL Containing Chlorinated Ethanes and Ethenes Using Hydrogen Peroxide and Persulfate in Soils[J]. Journal of Environmental Management, 2012, 108(16): 42-48. |
| [26] |
孟凡勇, 刘锐, 小林刚, 等. 挥发性氯代烃在湿润土壤中的平衡吸附研究[J].
环境科学, 2012, 33(4): 1361-1368.
Meng Fanyong, Liu Rui, Kobayashi Takeshi, et al. Study on Equilibrium Adsorption of Volatile Chlorinated Hydrocarbons on Humid Soils[J]. Environmental Science, 2012, 33(4): 1361-1368. |
| [27] |
张凤君, 贾晗, 刘佳露, 等. 有机氯代烃在壤土中的吸附和解吸特性[J].
吉林大学学报(地球科学版), 2015, 45(5): 1515-1522.
Zhang Fengjun, Jia Han, Liu Jialu, et al. Sorption and Desorption of Chlorinated Hydrocarbons onto Loam Soil[J]. Journal of Jilin University (Earth Science Edition), 2015, 45(5): 1515-1522. |
| [28] | Liang Chenju, Lee I L, Hsu I Y, et al. Persulfate Oxidation of Trichloroethylene with and Without Iron Activation in Porous Media[J]. Chemosphere, 2008, 70(3): 426-35. DOI:10.1016/j.chemosphere.2007.06.077 |
| [29] |
顾小钢, 吕树光, 邱兆富, 等. 热活化过硫酸盐处理地下水中氯代烃的研究[C]//上海市化学化工学会2011年度学术年会论文集. 上海: 化学世界, 2011: 125-126.
Gu xiaogang, Lü shuguang, Qiu Zhaofu, et al. Study on the Treatment of Chlorinated Hydrocarbons in Groundwater by Heat Activated Persulfate[C]//Shanghai Society of Chemistry & Chemical lndustry, a Collection of Annual Academic Annual Meetings for 2011. Shanghai: Chemical World, 2011: 125-126. |
| [30] | Chen Xiaoqing, Muthu M, Zhang Yanrong. Degrada-tion of p-Nitrophenol by Thermally Activated Persulfate in Soil System[J]. Chemical Engineering Journal, 2016, 283: 1357-1365. DOI:10.1016/j.cej.2015.08.107 |
| [31] | He Yan, Xu Jianming, Wang Haizhen, et al. Detailed Sorption Isotherms of Pentachlorophenol on Soils and Its Correlation with Soil Properties[J]. Environmental Research, 2006, 101: 362-372. DOI:10.1016/j.envres.2006.01.002 |