


0 引言
二恶烷(1, 4-dioxane)被广泛用作氯代溶剂如1, 1, 1-三氯乙烷、三氯乙烯等的稳定剂及树脂、植物油、矿物油和蜡等的溶剂[1]。其被国际癌症组织认定为2B级致癌物[2],世界卫生组织建议水体控制标准为50 μg/L。由于二恶烷水溶性强,加之传统检测方法的限制,上个世纪其地下水污染的问题并未受到广泛关注。随着2005年、2006年其检测方法的建立与完善,美国等发达国家在水源水、工业废水、溪水及地下水中大量检出二恶烷污染物,据美国超级基金统计,20%以上的氯代溶剂污染地下水中会检测到二恶烷存在[1, 3]。因此,近年来二恶烷被定为新型污染物质,其在环境中的行为、修复技术等得到环境污染治理领域的广泛关注。
二恶烷因其具有结构稳定,与水混溶及挥发性、吸附性能差等特点[1],在地下水中迁移速度快,较易造成大面积的污染[4],吸附、空气吹脱、化学还原等修复技术并不能有效将其去除[5-6],微生物也难以直接降解二恶烷,通常要加入共代谢基质将其降解[7]。高级氧化工艺是一种广泛应用于污水处理及场地修复的成熟技术,其处理速度较快,可有效将有机污染物氧化降解。目前已有利用电氧化、臭氧及光降解等方法处理二恶烷的报道[8-10]。
芬顿法以Fe2+活化H2O2产生羟基自由基(·OH)(氧化还原电位E0=2.80 V),·OH非常活泼,能够氧化降解目标污染物。然而在实际应用中,芬顿法会受H2O2消耗过快和pH变化等因素的限制[11]。过硫酸盐(PS,S2O42-)(E0=2.01 V)具有室温下溶解度高和稳定性强等特点[12],在加热、紫外光或过渡金属离子等条件的活化作用下,产生硫酸根自由基(SO4·-,E0=2.6 V),SO4·-较·OH具有更强的氧化性和非选择性,可有效降解染料、抗生素和化工原料与产物等污染物[13-15]。
有研究[16-17]表明,Fe2+是PS生成SO4·-的有效活化剂,Fe2+/PS体系可达到快速降解目标有机污染物的目的,零价铁(ZVI,Fe0)催化PS去除污染物的过程被认为是一种非均质反应,与Fe2+等过渡金属的均质反应相比,具有低价、pH适用广泛及具可回收性能等优点。PS与ZVI反应产生Fe2+,进而Fe2+活化PS生成SO4·-,反应式为:
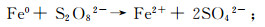 (1)
(1) 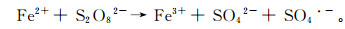 (2)
(2) 除反应式(1)外,ZVI在水中可分别与氧气、水以及Fe3+反应生成Fe2+,反应式为:
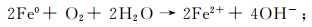 (3)
(3) 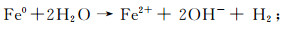 (4)
(4) 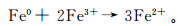 (5)
(5) ZVI/PS体系可有效氧化降解有机污染物包括一些难降解有机污染物,如2, 4-二硝基甲苯和对氯苯胺等[18-20];另外,ZVI在反应过程中可消耗溶液中Fe3+,同Fe2+催化体系相比较,可在一定程度上减少氢氧化铁沉淀的产生,进而减少材料的消耗。
除此之外,Weng等[21]和Du等[22]研究表明,在ZVI/PS体系中引入超声,可促进硫酸根自由基的产生,强化化工废水和染料的降解效果。由于二恶烷属于难降解有机污染物,因此本研究拟采用超声强化ZVI活化PS体系进行二恶烷污染地下水处理的实验研究,在对比超声强化作用的基础上,考察ZVI质量浓度、PS浓度以及碳酸根浓度等参数对二恶烷降解效果的影响,并对ZVI处理前后表面铁氧化合物组成进行对比分析,探索处理机理,以期为二恶烷污染地下水的高级氧化处理提供参考。
1 实验方法及材料 1.1 材料及药品二恶烷(1, 4-dioxane), ACS级(美国化学学会标准),购自百灵威公司;二氯甲烷(CH2Cl2)为色谱纯试剂,购自TEDIA公司;过硫酸钠(Na2S2O4)为分析纯试剂,购自天津光复精细化工研究所;ZVI聚集体粒径为0.297~2.380 mm,密度为2 240~2 560 kg/m3,购自美国Connelly-GPM公司;分析Fe2+质量浓度所用药剂购自美国哈希公司。地下水采自长春市,其碳酸根浓度为1.32 mmol/L,pH为7.16。
1.2 实验方法实验反应装置由超声仪、超声处理室、磁力搅拌器组成。在超声强化条件下,利用ZVI活化PS生成SO4·-降解二恶烷。实验中超声波恒定频率40 kHz,超声功率为100 W,并设置为脉冲超声模式,每间隔10 s超声5 s。二恶烷初始质量浓度为100 mg/L,PS浓度为5 mmo/L,ZVI质量浓度为2.0 g/L。按照一定间隔进行取样,样品经0.22 μm过滤器过滤后进行pH值、Fe2+质量浓度和二恶烷质量浓度分析。实验分别考察了不同初始PS浓度(1,5和10 mmol/L)、ZVI质量浓度(0.2,1.0和5.0 g/L)以及碳酸根浓度(1.32,2.00,3.00和5.00 mmol/L)对二恶烷降解效果的影响。
1.3 样品分析二恶烷样品前处理:取10 mL样品置于50 mL分液漏斗中,准确加入10 mL二氯甲烷,震荡10 min后静置5 min,移取1 mL有机相于气相色谱进样瓶中,待测。二恶烷测试方法:所用设备为气相色谱仪(美国安捷伦6890)分析色谱柱(Agilent 125-7032, DB-WAX, 30 m×530 μm×1 μm),色谱分析条件为进样口温度230 ℃、FID检测器温度250 ℃、载气流量5 mL/min。设置柱温升温程序: 40 ℃保持3 min,以10 ℃/min速度升温至100 ℃保持6 min。
Fe2+质量浓度采用分光光度计(美国哈希DR3900)进行检测,pH值使用便携式pH计(哈希HQ30D)进行测试。采用X射线光电子能谱分析(XPS)(美国Thermo公司,ES-CALAB250)及扫描电子显微镜(SEM-EDS)(日本SHIMADZU公司,SSX-550)对反应前后的ZVI进行表征分析。XPS测试条件为:Al K-α阳极,功率为200 W,全扫描透过能为150 eV,步长0. 5 eV,窄扫描透过能为60 eV,步长0. 05 eV,基础真空为10- 7 Pa。以C1s (284.6 eV)为定标标准进行校正。扫描电子显微镜测试方法为:取ZVI样品放置在载物板上,喷金后放入电子显微镜中进行分析。样品表面元素分析在200倍放大倍率下,选取500 μm×500 μm的面积进行能谱扫描。
2 结果与讨论 2.1 ZVI表征采用SEM-EDS分析样品处理前后ZVI表面的元素组成,其结果如表 1所示:Fe、O和C是ZVI表面的主要元素,占元素原子分数分别为41.84%、20.24%和36.45%。空气中的O2可将Fe氧化,使O元素以Fe的氧化物形态存在于ZVI表面;C元素则是工业生产Fe中的常见元素[23]。反应前后的对比表明,Fe原子分数下降18.25%,O元素提升22.08%。同时利用XPS对反应前后ZVI表面元素的化学形态进行分析,图谱如图 1所示,Fe元素的光谱由6条拟合曲线组成。其中,反应前(图 1a):Fe3O4对应的Fe3O4 2p3/2和Fe3O4 2p1/2的XPS峰,结合能分别为708.9和723.9 eV;Fe2O3对应Fe2O32p3/2和Fe2O32p1/2的XPS峰,结合能分别为712.8和726.5 eV,且分别在718.6和732.1 eV处具有卫星峰,这与文献[22]一致。对铁氧化物不同形态分析可知,反应前Fe3O4与Fe2O3的质量分数比为34.22%:65.77%,反应后质量分数比变为27.53%:72.46%,Fe3O4的质量分数比有所降低,说明一部分铁氧化物中Fe(Ⅱ)被转化为Fe(Ⅲ)。这主要是因为在酸性环境中ZVI表面铁氧化物中Fe(Ⅱ)被消耗,用于激活PS,同时Fe(Ⅲ)促进了形成Fe(Ⅱ)的铁循环;另外反应过程中产生的Fe(OH)3附着在ZVI表面,脱水后也可形成Fe2O3。
| 化学元素 | 原子分数/% | 质量分数/% | |||
| 反应前 | 反应后 | 反应前 | 反应后 | ||
| Fe | 41.84 | 23.59 | 74.05 | 53.64 | |
| O | 20.24 | 42.32 | 10.26 | 27.57 | |
| C | 36.45 | 31.34 | 13.87 | 15.33 | |
| S | 0.88 | 2.39 | 0.78 | 2.09 | |
| 其他 | 0.59 | 0.36 | 1.04 | 1.37 | |

|
| a.反应前,w(Fe3O4):w(Fe2O3)=34.22%:65.77%;b.反应后,w(Fe3O4):w(Fe2O3)=27.53%:72.46%。 图 1 ZVI的XPS图谱 Figure 1 XPS spectra of of iron |
|
|
实验分别设置了ZVI-PS和US-ZVI-PS两个体系进行二恶烷降解效果实验,对US(超声)、PS、ZVI、US-PS、US-ZVI进行空白对照(图 2),同时对ZVI-PS和US-ZVI-PS两个体系反应过程中的pH值和Fe2+质量浓度变化进行了监测(图 3)。结果表明:在仅有US和PS作用下,二恶烷的降解率分别为2.6%和3.8%,而仅有ZVI体系中二恶烷的降解率为11.0%,这主要是由于ZVI可以活化水中溶解氧并形成带有自由基的活性氧降解有机污染物[22-24];在US-ZVI体系中,二恶烷的降解率也为11.0%,说明US未对ZVI系统的氧化降解起到强化作用;在US-PS体系中二恶烷的降解率达到15.0%,降解率稍有提高的原因是在超声作用下,PS能够被超声活化产生SO4·-,如反应式(6),进而降解二恶烷。
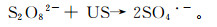 (6)
(6)

|
| ρ0,ρt分别为二恶烷的初始和t时刻的质量浓度(mg/L);t为时间(min)。 图 2 不同体系中二恶烷的降解 Figure 2 Degradation of dioxane in different systems |
|
|

|
| 图 3 超声对中Fe2+质量浓度的影响以及pH的变化 Figure 3 Effect ofultrasound on ferrous concentration and pH |
|
|
在ZVI-PS和US-ZVI-PS体系中,反应初始二恶烷的质量浓度迅速降低,之后伴随着一个缓慢降低的过程,在反应240 min时降解率分别达到85.0%和93.0%。ZVI-PS体系在60 min时二恶烷降解率为82.0%,而US-ZVI-PS体系在30 min就达到了80.0%。这表明超声对ZVI-PS氧化降解二恶烷起到了强化作用,一方面,通过式(6)途径产生了SO4·-;另一方面,超声可促进Fe2+的生成[25],如式(7)所示,增加了溶液中Fe2+质量浓度,进而加速了二恶烷氧化降解。
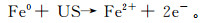 (7)
(7) 此外,超声对ZVI表面的冲击可提高物相间的传质效率。
由图 3可知:在ZVI-PS和US-ZVI-PS体系反应过程中,Fe2+质量浓度在90 min左右达到最大值,并趋于稳定,此时ZVI-PS体系中Fe2+的质量浓度为16.5 mg/L, US-ZVI-PS系统中Fe2+的质量浓度为30.0 mg/L;ZVI-PS系统和US-ZVI-PS系统中pH先由6.5降低至3.0左右,在30 min后上升,至90 min后趋于稳定,这与Fe2+质量浓度趋于稳定的时间一致。随着PS被活化的过程会产生H+,此过程反应式[18]为:
 (8)
(8) 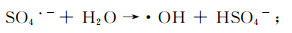 (9)
(9) 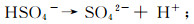 (10)
(10) 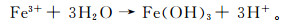 (11)
(11) 在反应初期,随着PS的消耗,pH迅速降低,当反应继续进行,在消耗ZVI的同时伴随着OH-的产生,如反应式(3)和(4)所示;因此,在60 min后pH上升,直至90 min Fe2+质量浓度达到稳定。由于在pH高于4.0的环境下,溶液中产生了铁沉淀物Fe(OH)2和Fe(OH)3,抑制了SO4·-的生成[26],因此二恶烷的降解速率降低。
2.3 PS浓度和ZVI添加量的影响实验考察了在US-ZVI-PS体系中不同PS浓度和ZVI质量浓度对二恶烷降解效果的影响,结果如图 4和5所示。SO4·-主要通过反应式(2)和(6)两个途径产生,因此高浓度的PS会产生更多的SO4·-,从而提升二恶烷的降解率。图 4中的结果与预期相同,在30 min时,10 mmol/L PS体系二恶烷降解率最高达到96.0%,而1 mmol/L的体系仅为5.0%。

|
| 图 4 过硫酸钠初始浓度对降解效率的影响 Figure 4 Effect of initial PS concentration on degradation efficiency |
|
|

|
| 图 5 ZVI初始质量浓度对降解效率的影响 Figure 5 Effect of ZVI Addition on degradation efficiency |
|
|
不同ZVI投加量对二恶烷处理影响的实验结果如图 5所示。当ZVI质量浓度为0.2 g/L时,二恶烷降解率最高,240 min降解率为99.6%。该体系在90 min前二恶烷降解率不足5%,从90 min开始二恶烷质量浓度迅速下降,最终接近完全降解。添加质量浓度为1.0 g/L和2.0 g/L的ZVI体系分别在80 min和60 min达到80%的降解率,在此后降解速度缓慢,240 min降解率达到90%左右,低于ZVI质量浓度为0.2 g/L体系的降解率。这一现象是由较高的Fe2+质量浓度所导致的。如反应式(12)所示,当Fe2+相对于SO4·-的质量浓度过量时,会产生Fe2+SO4·-竞争性消耗的现象[27]。
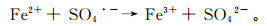 (12)
(12) 因此溶液中SO4·-浓度较低时,ZVI质量浓度为1.0 g/L和2.0 g/L的体系中高质量浓度的Fe2+会消耗SO4·-,降低了二恶烷的降解率。
2.4 碳酸根浓度的影响CO32-是地下水中水体中常见的阴离子,不同地区、季节的地下水碳酸根浓度不同,其浓度一般不超过10.00 mmol/L,因此本实验设置了1.32, 2.00, 3.00, 5.00 mmol/L几个不同碳酸根浓度,考察其对处理效果的影响。结果如图 6所示,随着CO32-浓度的增加,二恶烷降解效率逐渐降低,4个体系中二恶烷降解率分别为93%,86%,66%,42%。,这主要是因为CO32-的氧化还原电位(1.63 V)低于SO4·-的氧化还原电位,CO32-和HCO3-的存在会消耗SO4·-自由基(如反应式(13)和(14))[28],因此CO32-的浓度越高,污染物的降解效率越低。
 (13)
(13) 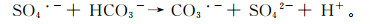 (14)
(14)

|
| 图 6 不同CO32-浓度对二恶烷降解的影响 Figure 6 Effect of CO32- concentration on degradation efficiency |
|
|
此外,CO32-和HCO3-还会与铁离子形成络合物或固体沉淀物,降低游离状态Fe2+的质量浓度[29]。因此在较高CO32-浓度的环境中,需要通过调节pH值等方式降低CO32-浓度,以降低对氧化反应的影响。
3 结论1) 超声一方面促进了Fe2+的产生,另一方面也促进了S2O42-转化为SO4·-,从而提高了氧化降解二恶烷的效果。
2) ZVI质量浓度和PS浓度的提高可以加快二恶烷的降解速率,然而当ZVI相对PS过多时,则会出现Fe2+抑制反应的现象。从降解效率和实现成本的角度考虑,ZVI的最佳质量浓度为1.0 g/L, PS的最佳浓度为10 mmol/L。
3) CO32-的存在会降低二恶烷降解效率,其负面作用主要体现在清除自由基和形成络合物沉淀物。
4) 超声强化ZVI活化PS可有效氧化去除地下水中污染物二恶烷。
| [1] |
Mohr T K, Stickney J A, Diguiseppi W H. Environ-mental Investigation and Remediation:1, 4-Dioxane and Other Solvent Stabilizers[M]. New York: CRC Press, Taylor & Francis Group, 2016.
|
| [2] |
Otto M, Nagaraja S. Treatment Technologies for 1, 4-Dioxane:Fundamentals and Field Applications[J]. Remediation Journal, 2007, 17(3): 81-88. DOI:10.1002/(ISSN)1520-6831 |
| [3] |
Zenker M J, Borden R C, Barlaz M A. Occurrence and Treatment of 1, 4-Dioxane in Aqueous Environments[J]. Environmental Engineering Science, 2003, 20(5): 423-432. DOI:10.1089/109287503768335913 |
| [4] |
Adamson D T, De Blanc P C, Farhat S K, et al. Implications of Matrix Diffusion on 1, 4-Dioxane Persistence at Contaminated Groundwater Sites[J]. Science of the Total Environment, 2016, 562: 98-107. DOI:10.1016/j.scitotenv.2016.03.211 |
| [5] |
Adams C D, Scanlan P A, Secrist N D. Oxidation and Biodegrability Enhancement of 1, 4-Dioxane Using Hydrogen Peroxide and Ozone[J]. Environmental Science & Technology, 1994, 28(11): 1812-1818. |
| [6] |
Lesage S, Jackson R E, Priddle M W, et al. Occu-rrence and Fate of Organic Solvent Residues in Anoxic Groundwater at the Gloucester Landfill, Canada[J]. Environmental Science & Technology, 1990, 24(4): 559-566. |
| [7] |
Lee I S, Sim W J, Kim C W, et al. Characteristic Occurrence Patterns of Micropollutants andTheir Removal Efficiencies in Industrial Wastewater Treatment Plants[J]. Journal of Environmental Monitoring, 2011, 13(2): 391-397. DOI:10.1039/C0EM00130A |
| [8] |
Barndok H, Hermosilla D, Cortijo L, et al. Elec-trooxidation of Industrial Wastewater Containing 1, 4-Dioxane in the Presence of Different Salts[J]. Environmental Science and Pollution Research, 2014, 21(8): 5701-5712. DOI:10.1007/s11356-013-2483-2 |
| [9] |
Suh J H, Mohseni M. A Study on the Relationship Between Biodegradability Enhancement and Oxidation of 1, 4-Oioxane Using Ozone and Hydrogen Peroxide[J]. Water Research, 2004, 38(10): 2596-2604. DOI:10.1016/j.watres.2004.03.002 |
| [10] |
Maurino V, Calza P, Minero C, et al. Light-Assisted 1, 4-Dioxane Degradation[J]. Chemosphere, 1997, 35(11): 2675-2688. DOI:10.1016/S0045-6535(97)00322-6 |
| [11] |
Hori H, Yamamoto A, Hayakawa E, et al. Efficient Decomposition of Environmentally Persistent Perfluoro Carboxylic Acids by Use of Persulfate as a Photochemical Oxidant[J]. Environmental Science & Technology, 2005, 39(7): 2383-2388. |
| [12] |
Criquet J, Leitner N K V. Degradation of Acetic Acid with Sulfate Radical Generated by Persulfate Ions Photolysis[J]. Chemosphere, 2009, 77(2): 194-200. DOI:10.1016/j.chemosphere.2009.07.040 |
| [13] |
Rodriguez S, VasquezA L, Costa D, et al. Oxidation of Orange G by Persulfate Activated by Fe (Ⅱ), Fe (Ⅲ) and Zero Valent Iron (ZVI)[J]. Chemosphere, 2014, 101: 86-92. DOI:10.1016/j.chemosphere.2013.12.037 |
| [14] |
Jafari A J, Kakavandi B, Jaafarzaneh N, et al. Fen-ton-Like Catalytic Oxidation of Tetracycline by AC@Fe3O4 as a Heterogeneous Persulfate Activator:Adsorption and Degradation Studies[J]. Journal of Industrial and Engineering Chemistry, 2017, 45: 323-333. DOI:10.1016/j.jiec.2016.09.044 |
| [15] |
Zhang Y, Tran H P, Du X, et al. Efficient Pyrite Activating Persulfate Process for Degradation of p-Chloroaniline in Aqueous Systems:A Mechanistic Study[J]. Chemical Engineering Journal, 2017, 308: 1112-1119. DOI:10.1016/j.cej.2016.09.104 |
| [16] |
Liang C, Bruell C J, Marley M C, et al. Persulfate Oxidation for in Situ Remediation of TCE:Ⅰ:Activated by Ferrous Ion with and Without a Persulfate-Thiosulfate Redox Couple[J]. Chemosphere, 2004, 55(9): 1213-1223. DOI:10.1016/j.chemosphere.2004.01.029 |
| [17] |
Rao Y, Qu L, Yang H, et al. Degradation of Car-bamazepine by Fe (Ⅱ)-Activated Persulfate Process[J]. Journal of Hazardous Materials, 2014, 268: 23-32. DOI:10.1016/j.jhazmat.2014.01.010 |
| [18] |
刘娜, 王柳, 邱华, 等. 生物炭催化过硫酸盐脱色偶氮染料金橙Ⅱ[J]. 吉林大学学报(地球科学版), 2014, 44(6): 2000-2009. Liu Na, Wang Liu, Qiu Hua, et al. Biochar-Catalyzed Persulfate Decolorization of Azo Dye Gold Orange Ⅱ[J]. Journal of Jilin University (Earth Science Edition), 2014, 44(6): 2000-2009. |
| [19] |
Hussain I, Zhang Y, Huang S, et al. Degradation of p-Chloroaniline by Persulfate Activated with Zero-Valent Iron[J]. Chemical Engineering Journal, 2012, 203: 269-276. DOI:10.1016/j.cej.2012.06.120 |
| [20] |
Santos-Juanes L, Einschlag F G, Amat A, et al. Combining ZVI Reduction with Photo-Fenton Process for the Removal of Persistent Pollutants[J]. Chemical Engineering Journal, 2017, 310: 484-490. DOI:10.1016/j.cej.2016.04.114 |
| [21] |
Weng C H, Lin Y T, Yuan H M. Rapid Decoloration of Reactive Black 5 by an Advanced Fenton Process in Conjunction with Ultrasound[J]. Separation and Purification Technology, 2013, 117: 75-82. DOI:10.1016/j.seppur.2013.03.047 |
| [22] |
Du J, Bao J, Fu X, et al. Mesoporous Sulfur-Modified Iron Oxide as an Effective Fenton-Like Catalyst for Degradation of Bisphenol A[J]. Applied Catalysis B:Environmental, 2016, 184: 132-141. DOI:10.1016/j.apcatb.2015.11.015 |
| [23] |
Son H S, Im J K, Zoh K D. A Fenton-Like Degradation Mechanism for 1, 4-Dioxane Using Zero-Valent Iron (Fe 0) and UV Light[J]. Water Research, 2009, 43(5): 1457-1463. DOI:10.1016/j.watres.2008.12.029 |
| [24] |
Shin J, Lee Y-C, Ahn Y, et al. 1, 4-Dioxane Degradation by Oxidation and Sonication in the Presence of Different-Sized ZVI in Open-Air System[J]. Desalination and Water Treatment, 2012, 50(1/2/3): 102-114. |
| [25] |
Fu F, Lu J, Cheng Z, et al. Removal of Selenite by Zero-Valent Iron Combined with Ultrasound:Se (Ⅳ) Concentration Changes, Se(Ⅵ) Generation, and Reaction Mechanism[J]. Ultrasonics Sonochemistry, 2016, 29: 328-336. DOI:10.1016/j.ultsonch.2015.10.007 |
| [26] |
Wei X, Gao N, Li C, et al. Zero-valent Iron (ZVI) Activation of Persulfate (PS) for Oxidation of Bentazon in Water[J]. Chemical Engineering Journal, 2016, 285: 660-670. DOI:10.1016/j.cej.2015.08.120 |
| [27] |
Tan C, Gao N, Chu W, et al. Degradation of Diuron by Persulfate Activated with Ferrous Ion[J]. Separation and Purification Technology, 2012, 95: 44-48. DOI:10.1016/j.seppur.2012.04.012 |
| [28] |
Asghar A, Raman A A A, Daud W M A W. Advanced Oxidation Processes for In-Situ Production of Hydrogen Peroxide/Hydroxyl Radical for Textile Wastewater Treatment:A Review[J]. Journal of Cleaner Production, 2015, 87: 826-838. DOI:10.1016/j.jclepro.2014.09.010 |
| [29] |
Keen O S, Mckay G, Mezyk S P, et al. Identifying the Factors that Influence the Reactivity of Effluent Organic Matter with Hydroxyl Radicals[J]. Water Research, 2014, 50: 408-419. DOI:10.1016/j.watres.2013.10.049 |