 2020, Vol. 48
2020, Vol. 48文章信息
- 刘玉项, 朱胜, 韩冰源
- LIU Yu-xiang, ZHU Sheng, HAN Bing-yuan
- 金属镁电化学腐蚀阳极析氢行为研究进展
- Research progress in anodic hydrogen evolution of magnesium electrochemistry corrosion
- 材料工程, 2020, 48(10): 17-27
- Journal of Materials Engineering, 2020, 48(10): 17-27.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2019.001078
-
文章历史
- 收稿日期: 2019-11-19
- 修订日期: 2020-06-15




2. 江苏理工学院 汽车与交通工程学院, 江苏 常州 213001
2. School of Automobile and Traffic Engineering, Jiangsu University of Technology, Changzhou 213001, Jiangsu, China
轻量化优势[1]和优良的可降解能力[2]赋予了镁及其合金在国防、工业、医用等领域强大的应用动力。但是,在中性或酸性水溶液中较快的腐蚀速率和复杂的腐蚀机理极大地制约了它们的广泛应用。在金属镁的腐蚀过程中,存在着阳极析氢的特殊现象,其机理的揭示对于理解镁及其合金复杂的腐蚀过程以及改善其耐蚀性能至关重要。
金属镁复杂的腐蚀机理是由其高电化学活性、脆弱的腐蚀产物和特殊电化学行为决定的。首先,镁能在水溶液中自发地腐蚀溶解,其还原反应电位和电偶腐蚀电位均是较大的负值,导致了镁的高反应活性[3-4]。其次,镁的氧化膜在水中能够快速溶解,且腐蚀生成的产物只能在很窄的碱性pH范围内稳定存在[5-6]。最后,镁在极化时会出现阳极析氢现象。根据腐蚀电化学理论,金属在阳极极化过程中阴极反应会被抑制,阴极反应速率会随阳极极化电压的增大而减小[7-9]。但是,金属镁在阳极极化的过程中,仍然存在大量氢气释放,且释放量随阳极电压的增大而增加。这种特殊的现象被称为“负差数效应(negative difference effect,NDE)”[10-12],目前解释这种现象的假设和理论尚未完善。对于NDE现象,1866年德国化学家Beetz[13]首次在金属镁上发现了这种效应:当镁电极在含
金属镁的腐蚀是一个电化学过程。在水溶液中,纯镁电极上阳极区镁原子溶解,阴极区氢气析出。从热力学角度来说,镁电极与水溶液形成的腐蚀体系自由能差小于0,导致了镁能够自发地腐蚀溶解。从动力学角度来说,当电极上未施加过电位时,镁电极在水溶液中达到平衡时阳极反应速率等于阴极反应速率,此时的电位为平衡电位,对应的电流密度为交换电流密度。而当电极上施加过电位时,如果考虑放电步骤是速率控制步骤,镁阳极溶解反应和水阴极反应的过电位曲线都可用Butler-Volmer方程[8, 15]来表达:
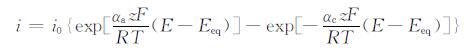
|
(1) |
式中:E是外部电压,V;i是在施加外部电压下所得电流密度,A/cm2;i0是交换电流密度,A/cm2;αa/αc是阳极/阴极的传递系数;z是电化学反应转移的电子数;F是法拉第常数,96485.34 C/mol;R是气体常数,8.314 J/(K·mol);T是开氏温度,K;Eeq是平衡电位,V;E-Eeq是过电位,V。
当镁在水溶液中发生腐蚀溶解时,镁电极上同时有两个电极反应,因为腐蚀电位离这两个电极反应的平衡电位都比较远,当施加电压E=Ecorr时,镁原子溶解的阳极反应可忽略式(1)右侧第二部分,而氢气析出的阴极反应可忽略式(1)右侧第一部分。根据极化曲线合成原理,整个金属电极的外测电流密度与施加电压的关系可用E-I曲线表达[8]:
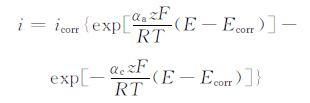
|
(2) |
式中:icorr是金属的平均腐蚀电流密度,A/cm2;Ecorr是腐蚀电位,V。由式(2)可知,当阳极极化(E-Ecorr
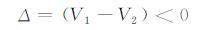
|
(3) |
式中:V1是镁自由腐蚀时阴极反应的氢气释放量;V2是镁阳极极化时阴极反应的氢气释放量;Δ为二者之差。Δ<0意味着阳极极化时镁电极会表现出“负”的氢气释放量的差数效应。图 1展示了根据式(2)绘制的表征纯镁“负差数效应”现象的极化曲线[11]。

图 1中,ia和ic分别是纯镁电化学腐蚀的阳极、阴极反应Tafel外插线;icorr是镁平均腐蚀电流密度,当E=Ecorr时,icorr=ia=ic;iMg和iH分别为活性控制极化时代表纯镁溶解和代表氢气释放的腐蚀电流密度。当施加阳极电压Eappl时,平行于横坐标的直线上的iH, e,iH, m,iMg, e和iMg, m分别是根据式(2)计算的阴极反应电流密度,电极上实际氢气释放的电流密度,阳极反应电流密度和代表实际镁溶解的电流密度。其中,Δ1=iMg, e-iH, e是“正差数效应”的金属(如Fe,Cu等)阳极极化的腐蚀电流密度,即式(2)的i。Δ2=i0-iH, m<0是和式(3)等价的NDE表达式。Δ3=iH, m-iH, e>0表明了阳极极化时纯镁电极上实际氢气释放速率远超过理论氢气释放速率。Δ4=iMg, m-iMg, e>0表明了实际纯镁电极腐蚀溶解的电流密度要大于由恒电位仪记录的阳极腐蚀电流密度,即阳极极化时纯镁实际溶解导致的电流密度超过法拉第电流密度。因此,对于存在NDE现象的金属,如果采用Tafel方法计算腐蚀电流密度,则其值小于实际氢气释放和实际电极溶解对应的总的腐蚀电流密度[16]。
此外,由图 1可知,iMg随着阳极电压增大呈指数增大,而iH随着阳极电压增大呈线性增大。但是,iH与阳极电压之间并非是严格的线性关系,实际上,在一定的阳极过电位范围内,iH是正常的“正差数效应”。因此,NDE的表述并不十分准确。目前更多研究者把NDE现象更严格地称为“反常氢气释放”或“额外阳极氢气释放”[12, 15-17],以避免和阴极反应中的氢气释放混淆。为了方便和统一,本文沿用NDE代指金属镁腐蚀的阳极析氢现象。
2 金属镁电化学腐蚀阳极析氢行为机理自金属镁阳极极化NDE现象被发现以来,研究者提出了大量的假设,建立了各种模型并采用理论和实验结合的方法试图揭示NDE的机理,表 1总结了揭示金属镁腐蚀NDE现象机理最常见的几种假设。
| Hypothesis | Ref | Comment |
| Univalent Mg | [11, 18] | Being criticized |
| Enhanced cathodic activity of corrosion products | [16, 19-22] | Plays a role |
| Enrichment, release and redeposit of impurities | [23-25] | Plays a role |
| Chunking (non-faradaic loss of Mg) | [13, 26] | No strong evidence |
| Remote current model | [19, 27] | Plays a role, needs further study |
| Catalytic nature of dissolving surface | [28-30] | Potentially plays a dominate role |
单价态Mg离子理论假设Mg+存在,其作为介于Mg原子和Mg2+离子的中间价态参与镁电极的阳极反应,能提供更多的阳极反应步骤。Song等[11, 18]认为镁的阳极反应分两步:电化学反应中一部分Mg原子被氧化形成Mg+,释放一部分H2,见式(4);Mg+再次发生均衡的化学反应,生成Mg2+和H2,见式(5)。

|
(4) |

|
(5) |
由式(4),(5)可知,施加阳极电压时,式(4)反应加快,从而Mg+进入溶液的速度加快,导致了式(5)中反应加速,从而氢气释放加速。
这个理论的根基在于假设Mg+的存在。Gomberg[30]在1929年首先假设在乙醚苯溶液镁的卤化物中存在单价Mg+;随后,Petty等[31]在1954年研究金属镁腐蚀NDE现象时沿用了Mg+存在的假设。他们首先开展了
因此,根据式(4),(5),单价态Mg离子理论能够解释NDE现象,而且自从这个理论提出以来,由于一些实验结果和该理论相吻合,后续大量研究对该理论进行了引用,成为揭示金属镁“负差数效应”的最广为接受的理论之一。但是,如表 1所评论的,因为没有Mg+存在的实验证据[33],目前这个理论备受质疑。另一方面,目前实验证明镁阳极反应中氧化价态总是等于2,不存在低于+2的Mg离子。例如,Rossrucker等[34]采用原位感应耦合等离子发射谱测定了Mg在流动的NaCl溶液腐蚀体系中腐蚀后的离子浓度,化学计量结果显示镁氧化后仅以+2价离子存在。相似地,Swiatowska等[35]采用原子发射光谱电化学研究了镁在NaCl和Na2SO4溶液中的阳极溶解,结果表明只有+2价Mg离子参与电化学反应。总之,除非在后续研究中实验证实Mg+的存在,否则单价态Mg离子理论只能作为一个纯假设而不能作为揭示NDE机理的主导理论。
2.2 高活性膜层理论纯镁的腐蚀产物包含双层的MgO/Mg(OH)2薄膜,肉眼观察呈黑色。这层黑色膜层被认为具有提高金属镁阴极反应活性的能力[16, 19-22]。近年来研究发现,镁自由腐蚀时,相比尚未被腐蚀的区域,被黑色膜层(腐蚀产物)覆盖的区域上氢气释放的速率更快。Williams等[36]采用原位扫描振动电极(scanning vibration electrode technique, SVET)系统开展了纯镁在5%(质量分数,下同)NaCl溶液中自由腐蚀时表面黑色膜层对阴极活性的影响实验。他们发现,黑色膜层区域局部的阴极活性很高;同时,由SVET得到的腐蚀电流密度以及由氢气收集法和腐蚀失重法得到的腐蚀速率均受到高阴极活性面积的影响。相似地,Curioni[16]采用恒电位仪记录了纯镁电极在3.5% NaCl溶液中极化时氢气释放量随过电位的变化趋势。他们发现,相比未腐蚀区域,腐蚀区域的阴极活性显著增强。此外,腐蚀后黑色薄膜逐渐向未发生腐蚀的区域扩张,在此过程中,氢气释放量逐渐增加,且释放速率最快区域集中在腐蚀前沿(腐蚀区域与未腐蚀区域的边界)。腐蚀前沿具有较高的阴极反应活性,加快了此处镁阳极溶解的速率,从而增强了腐蚀扩展的动力。
基于黑色膜层提高阴极反应活性的实验结果,Williams等[36]建立了如图 2所示纯镁腐蚀模型。图中,A代表阳极反应过程,C代表阴极反应过程。在腐蚀区域不断向未腐蚀区域扩张的过程中,腐蚀前沿处镁阳极溶解产生的电子,一部分被黑色膜层处H2O分子获取,产生H2;另一部分转移到恒电流仪中被记录为腐蚀电流密度。由此可知,在纯镁自由腐蚀时,恒电位仪(或恒电流仪)记录的腐蚀电流密度小于实际镁溶解导致的阳极反应速率。因此,黑色膜层促进阴极活化效应能够导致NDE现象中额外的氢气释放及更多的镁阳极溶解。

此外,Birbilis等[37]开展了一系列实验来验证膜层理论。他们观察了纯镁电极在0.1 mol/L NaCl溶液中被施加2.5 mA/cm2和20 mA/cm2的电流2 min后的表面形貌,如图 3所示。由图可知,在阳极极化时,腐蚀产物导致的黑色膜层的面积随阳极电压的增大而增大。显然,相同腐蚀时间内,相比自由腐蚀,在阳极极化时腐蚀前沿面积更大,从而Mg溶解释放电子速率更快,在更大面积的黑色膜层上氢气释放量更多,而恒电流仪能够记录的腐蚀电流密度相对变小。此外,他们还对纯镁进行阳极极化,然后测量阴极极化下的析氢电流的实验,发现阳极极化的电流越大,黑色膜层面积越大(图 3(b)),导致测量到的析氢电流也越大。他们还发现即使在弱阳极极化下,随时间延长,黑色膜层面积逐渐增大,会因此导致电流从阳极转化成阴极,出现“极性反转”的现象。

|
图 3 纯镁在0.1 mol/L NaCl中施加不同阳极电流时黑色膜层变化[37] (a)施加2.5 mA/cm2阳极电流2 min;(b)施加20 mA/cm2阳极电流2 min Fig. 3 Growth of the dark regions upon pure Mg during anodic polarisation in 0.1 mol/L NaCl[37] (a)Mg after application of an impressed current of 2.5 mA/cm2 for 2 min; (b)Mg after application of an impressed current of 20 mA/cm2 for 2 min |
上述实验都证明了膜层理论能够一定程度解释NDE现象。实际上,高阴极活性膜层理论是基于由Frankel等[38]提出的析氢交换电流i0增大的机理,他们认为镁腐蚀后产生的高活性膜层的析氢交换电流大于未腐蚀镁基体的析氢交换电流。因此,根据式(1)可得到Tafel方程式(6):
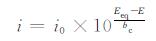
|
(6) |
式中:bc为阴极Tafel斜率。由式(6)可知,如果黑色膜层导致了纯镁电极的析氢交换电流i0增大,那么在阳极极化时,析氢速率增大。此外,因为黑色膜层的结构、成分以及在NDE现象中发挥的作用可以用实验手段获得,而且实验结果证明膜层确实在镁阳极极化时起到促进氢气释放的作用,因此高活性膜层理论近年来受到越来越多关注,目前快速发展的实验手段和方法促进了该理论的逐渐成熟。
2.3 杂质/合金原子富集效应镁的腐蚀产物不仅是由MgO/Mg(OH)2双层膜组成,还含有富集的杂质/合金团簇原子。例如,Taheri等[23]采用高角环形暗场扫描透射电子显微镜观察到镁腐蚀后黑色膜层中有Fe杂质原子的富集。Gore等[39]采用粒子诱导X射线光谱(particle-induced X-ray spectroscopy,PIXE)研究了Mg-0.1%In和Mg-0.1%Au在0.1 mol/L NaCl溶液中阳极极化(2 mA/cm2和10 mA/cm2)2 min后腐蚀产物中In和Au的富集情况。图 4为In和Au的富集比随施加电荷密度变化规律。富集比是指腐蚀产物中合金元素的含量与基体中合金元素含量的比值。由图 4可知,阳极极化后腐蚀产物中两种合金元素均出现富集;Au的富集更加明显,当施加5 C/cm2的电荷密度时,Au的富集比达到70左右。因为这些杂质/合金元素具有比Mg更高的析氢交换电流,当它们在腐蚀产物中富集并接触腐蚀介质时,无论镁合金电极是否极化,这些富集的原子都会整体提高镁合金腐蚀速率。另一方面,杂质/合金元素与镁形成的二次相具有比α-Mg相更高的自腐蚀电位,从而促进镁的电化学反应活性。在α-Mg相作为阳极被优先腐蚀之后,作为阴极的二次相就会裸露在溶液中,增强了阴极反应活性并导致额外氢气的释放。

基于杂质/合金原子在腐蚀产物中的富集效应,Taheri等[23]建立了镁腐蚀NDE模型(以Fe原子富集为例),如图 5所示。阳极极化之前,Fe杂质原子富集在镁基体与MgO(在空气中形成)界面处,并未与溶液直接接触。此时,镁腐蚀产生的电子完全被H2O分子的还原反应消耗。阳极极化时,富集的Fe原子随腐蚀产物的生成迁移到电极/溶液表面,此时,Fe原子上H2O分子阴极反应加速,消耗了较多的电子,导致镁氧化产生电子的速率加快,但是能够被恒电位仪记录的电子数却减少,从而导致了NDE。

和杂质/合金原子富集效应类似,一些学者提出了镁原子分散溶解效应,他们认为在腐蚀产物中富集的并非是杂质/合金原子,而是Mg原子。Straumanis等[26]研究发现在自由腐蚀或阳极极化时,镁原子会脱离晶格变成精细的颗粒状或薄片状(实际是大量镁原子聚集体),这些细小的结构分散在Mg(OH)2腐蚀产物中,使腐蚀表面整体呈黑色。在施加较高的阳极电流后,黑色表面膜局部破裂,这些分散在腐蚀产物薄膜中的团簇镁原子接触到溶液后被氧化,导致了额外的阴极反应步骤。实际上,Marsh等[40]研究Fe的阳极析氢时提出过相似的假设,即在一定条件下,Fe的腐蚀一部分是通过含有一定数量Fe原子的金属碎片脱离金属表面之后进行的。当施加更高的阳极电压时,这种分散溶解效应更加明显。这种腐蚀过程金属原子脱离晶格并以颗粒状或片状的分散溶解现象也在Hoey等[41]的研究中提到。显然,当分散在腐蚀产物膜层中镁的团簇原子接触溶液发生阳极反应时,分散的镁原子溶解和氢气释放导致的电流难以用恒电位仪记录到,从而发生了“负差数效应”。但是,如表 1所示,镁原子团簇富集在腐蚀产物中产生镁阳极极化时的“非法拉第电流”并没有实验结果予以验证,实际上,镁原子分散溶解效应只是与杂质/合金原子富集效应相对应的一个假设,实验中并未观察到这一效应。例如,Taheri等[23]分析了镁腐蚀后黑色膜层的截面特征,没有发现疏松颗粒状或片状的Mg原子团簇。这个否定镁原子团簇存在的结论在文献[42-44]中得到证实。实际上,真正存在的颗粒状结构可能是杂质原子,而NDE现象则更可能是黑色膜层和杂质原子共同作用导致阴极反应活性的提高引起的。
2.5 “远端电流”模型上述膜层理论和金属原子富集理论有一个共同点,即存在使镁氧化产生更多电子的因素,且这些电子并未被恒电流仪完全记录,而是在电极表面某处被H2O分子发生的阴极反应消耗掉。基于这样的观点,Curioni等[27]提出“远端电流(remote current)”模型,他们认为腐蚀产物形成的膜层以及杂质/合金元素上阴极反应活性高,在其上的H2O分子的还原需要更多的电子,这些电子由纯镁的溶解提供,从而在镁电极上形成了“远端电流”。而且,随着阳极电压的升高,这些区域阴极反应活性越高,生成的H2越多,“远端电流”也越大,导致镁溶解的质量超过失重量的法拉第结果。
在Curioni的实验中,他们研究了纯镁(99.95%)在3.5% NaCl溶液中释放氢气的过程,建立了如图 6所示的镁腐蚀的“远端电流”模型。图中,在左侧未腐蚀区域(a区),富集的杂质原子(或二次相、金属间化合物等)接触溶液后,由于阴极活性强,能够快速释放H2气泡,逐渐聚集形成大气泡并吸附在富集点。大的气泡形成过程导致气泡附近溶液的碱性化,降低了富集区镁阳极溶解速率。该区域阴极反应消耗的电子形成电流,并流入腐蚀前沿(b区),作为一部分“远端电流”加快了腐蚀前沿的镁阳极溶解速率。此外,在右侧已腐蚀区域(c区),覆盖了腐蚀产物膜层,黑色膜层及其中裸露的杂质原子富集增强了此处阴极反应活性,作为另一部分“远端电流”加剧腐蚀前沿处镁的阳极溶解。在c区,氢气以小气泡形式溢出,局部pH值升高。由气泡和尚未沉淀的腐蚀产物形成了高pH的凝胶态[19],其也许会导致此处溶液电阻升高,发生欧姆极化。在腐蚀前沿,相对低的pH和高Cl-浓度导致此区域发生了极快的阴、阳极反应。因此,该区域镁溶解的阳极电流密度实际上是阳极“法拉第”电流密度与两部分“远端电流”导致的电流密度的总和。由此可知,镁腐蚀溶解的质量大于通过法拉第定律计算的质量损失。

值得注意的是,上述模型是在镁自然腐蚀状态下得到的,即是说“远端电流”模型表明NDE现象在镁自然腐蚀时也存在,并不需要在强阳极极化的条件下。而在强阳极极化时,除了两部分“远端电流”所需要的电子,恒电流仪也需要大量电子维持阳极极化电流,这些电子均需要由镁快速溶解提供。显然,此时镁腐蚀的动力更足,腐蚀前沿处镁溶解速率更快,并超过了阳极“法拉第”电流密度,导致了更加显著的镁腐蚀NDE现象。
“远端电流”模型解释了金属镁在非阴极极化状态下均会出现“负差数效应”,因此可以说阳极极化只是放大了自由腐蚀状态下的“负差数效应”,而该效应的机理的揭示最终在于更深入探讨金属镁自由腐蚀时腐蚀机理与经典理论的差别,构建金属镁独特的腐蚀理论。
2.6 其他假设和理论Perrault[45]曾经发表了一个改进的镁腐蚀Pourbaix图,在原有E-pH图上添加了Mg+和MgH2两种物质。他假设镁的腐蚀反应除了最终生成的Mg2+,OH-和H2外,还有MgH2作为中间物质参与化学反应。随后,MgH2在实验中被发现[46],其在水溶液中的反应方程式为:
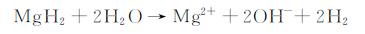
|
(7) |
显然,式(7)中MgH2可以像Mg+一样在镁阳极极化时促进氢气释放。但是,这种中间物质是否确实存在以及其稳定性仍然未知。
此外,Bender等[47]提出镁的腐蚀依赖于两个阴极过程,一个是来自于可以用恒电流仪测量的外部极化电流,另一个来自于能在腐蚀表面观察到氢气气泡的H+的富集效应导致的电流。而H原子的富集导致了阳极极化时额外的H2释放,由此产生NDE现象。
最近,Taylor[29]提出一种H2O分子在镁电极表面吸附和解吸附的模型,研究了镁在阴极、阳极极化时的电化学行为,并采用密度泛函理论计算了反应速率。根据该模型,镁腐蚀电化学反应为:
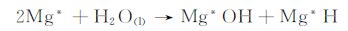
|
(8) |
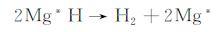
|
(9) |

|
(10) |

|
(11) |
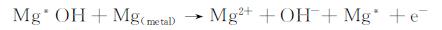
|
(12) |
式(8)揭示了镁表面吸附的H2O分子分解为H原子和羟基官能团(-OH)的过程,这个过程占据两个Mg*(Mg*是金属镁表面上一种金属态区域)金属单质。随后,两个H(依附于Mg*H)接触时结合生成H2,同时恢复两个Mg*金属单质,见式(9)。同时,-OH官能团(依附于Mg*OH)得到一个电子变成OH-,而Mg*金属恢复单质态,见式(10)。在阳极极化时,两个H2O分子吸附在4个Mg*金属单质上,得到电子被还原成H2,4个Mg*金属单质仅作为反应载体不发生氧化反应,见式(11);而对于正常的金属Mg单质,其发生氧化反应失去两个电子,供式(10), (11)的还原反应消耗,而-OH官能团依附的Mg*金属单质仍然不参与氧化还原反应,只作为反应载体,见式(12)。由式(8)~(12)可知,H2O分子在仅作为反应载体的Mg*金属单质表面的吸附和解吸附过程为NDE提供了更多的H原子,从而造成阳极析氢现象。
2.7 上述假设或理论的总结首先,对于依赖中间产物的理论,如Mg+,MgH2,H原子以及H2O分子吸附,这些理论成立的前提是中间产物存在并且参与镁的阳极极化。而实际上,目前并无确切的实验证据证明Mg+和MgH2的稳定存在,而H原子以及H2O分子的理论也无法通过实验证实,因此,这些假设无法完全揭示NDE现象的本质。
其次,对于膜层理论和金属原子富集效应,实验中也存在很多相互矛盾的情形。例如,膜层理论无法解释镁在酸性溶液中也存在的NDE现象;此外,尽管实验结果表明,在阳极极化时,腐蚀产物黑色膜层的面积随时间逐渐增大,相应地,氢气在更多的腐蚀前沿快速释放,且释放量不断增加。但是,通过计算镁极化过程随时间变化的实时氢气释放量可知,氢气释放速率是恒定值,并非随着阳极电压增加而加快,并不符合图 1中iH的变化趋势。此外,研究发现,杂质原子富集效率非常低,无法证明其与阴极活性提高之间的相关性。Birbilis等[48]通过PIXE技术得到了杂质原子在镁腐蚀产物中的富集效率小于1%的研究结论;Fajardo等[49]发现杂质元素含量<10-6 g/m3的高纯镁,腐蚀之后杂质原子的富集可以忽略,但是氢气释放速率仍然随阳极极化电压升高而加快。总之,黑色腐蚀产物膜层理论和杂质/合金原子富集效应均存在不足,它们在镁腐蚀NDE中起到一定作用,但是并非决定性因素。
Frankel等[28]同时使用原位扫描振动电极系统、恒电流仪、恒电位极化仪以及氢气收集装置研究了高纯镁(99.99%)在2 mol/L NaCl溶液中的腐蚀行为和NDE现象。根据研究结果,在阳极极化时,总氢气释放量对应的电流密度可表达为:
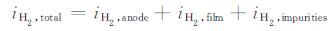
|
(13) |
式中:iH2, total是通过氢气收集装置获得的电流密度;iH2, anode是镁阳极溶解对应的电流密度;iH2, film和iH2, impurities分别是黑色膜层、杂质/合金元素富集导致阴极活化所得的电流密度。图 7为式(13)中各腐蚀电流密度随施加的阳极电流密度的变化曲线。由图可知,iH2, film和iH2, impurities对应的电流密度很小,且并不随着施加的阳极电流密度的增大而升高。因此,即使二者均能通过提高阴极反应活性增加阳极氢气的释放量,它们对镁腐蚀NDE现象也不起主导作用。实际上,图中二者所导致的腐蚀电流密度是几乎恒定的,这与NDE中氢气释放速率随阳极电压增大而加快相悖。

图 7中镁阳极溶解导致氢气释放的电流密度(iH2, anode)和氢气收集法得到的电流密度(iH2, total)相差不大,表明NDE现象实际上是由发生镁溶解的区域的阳极反应活性决定的,且该区域中阴极反应的活性随着施加电流的增大而升高[28-30]。
虽然目前仍没有完善的理论模型和实验证据揭示镁腐蚀的NDE现象,但研究者通过新的研究手段和方法获得了一些较为可靠的理论和解释,而最新的研究结果表明NDE现象本质上还是与镁阳极溶解的区域的活性密切相关。例如,Fajardo等[50-51]同时采用电化学测试和实时氢气收集,通过分析电化学手段加载电流密度和氢气释放输出电流密度之间的关系,结果否定了膜层理论和杂质/合金原子富集效应,但验证了Frankel等[28]提出的溶解区域活化的假设。这样的假设使NDE现象的研究回归到其本质,即阳极极化下氢气的释放由阳极溶解区域决定,其他因素如中间物质、合金化、杂质原子、腐蚀产物等可能只是放大了这种效应,并不起根本的作用。但是,为了建立完善的揭示该现象的理论,上述因素和一些更为关键的因素,如局部pH的大小与其所起的作用,溶液中溶解其他气体分子[52-53](如O2,CO2等),也亟须开展深入的研究。
3 结束语金属镁在水溶液中的阳极析氢行为作为镁电化学腐蚀的重要特征,其机理的揭示将会显著扩展镁及其合金作为结构和功能材料的应用领域。虽然基于腐蚀形貌观察和电化学测试以揭示NDE机理的研究取得了一些突出的成果,但要建立能够被研究者广泛接受的机理模型还依赖于更深入的研究:
(1) 近年来镁腐蚀阳极析氢现象的研究得到了新的研究方法(如第一性密度泛函理论[29, 54-55]和分子动力学模拟[54-56])和测试方法、技术(如氢气测量和电化学极化的结合方法[16, 51]以及原位电化学技术和原子发射光谱技术[57]结合)的推动,理论假设和实验验证均快速发展。采用原位电化学测试技术能够使局部腐蚀特征得到更加直观的展示。但是,对于影响局部电化学腐蚀特征的因素,包括局部pH,Cl-离子浓度,微电偶腐蚀体系以及局部溶液电阻的测量和研究,还有赖于更精确的测试探针的发明[12, 48]和微区电化学测试仪器和微电极[57-58]的使用。
(2) 虽然目前研究者针对镁腐蚀的NDE现象开展了大量研究,提出了很多假设和理论,但是多数研究者只是针对其中一个方面开展研究。例如,膜层理论中NDE机理研究集中在黑色腐蚀产物完全覆盖腐蚀电极表面之前,而对于被黑色膜层完全覆盖后的镁电极的NDE现象却鲜有分析,下一步研究的重点在于对黑色膜层完全覆盖镁电极后膜层理论是否可靠进行验证。此外,对于除了Mg之外的其他金属的NDE现象以及合金元素对NDE的影响规律也未深入研究。显然,通过对比不同金属和合金相似的NDE现象,更有利于建立完善的腐蚀理论。
(3) 鉴于金属腐蚀的研究都是以传统金属腐蚀电化学理论为基础,金属镁也不例外。但是,镁特殊的腐蚀电化学阳极析氢现象难以通过经典电化学理论解释。因此,考虑镁腐蚀过程中固/液界面双电层的电荷及物质传递的特殊模式,修正经典电化学的某些参数(例如电极反应的传递系数、离子的活度系数,气体释放导致的溶液黏度系数、密度等)或许能另辟蹊径,揭示镁腐蚀的NDE现象的机理。
| [1] |
CAROU D, DAVIM J P. Machining of light alloys:aluminum, titanium, and magnesium[M]. Florida: CRC Press, 2018.
|
| [2] |
ZHAO D W, WITTE F, LU F Q, et al. Current status on clinical applications of magnesium-based orthopaedic implants:a review from clinical translational perspective[J]. Biomaterials, 2017, 112: 287-302. DOI:10.1016/j.biomaterials.2016.10.017 |
| [3] |
程英亮, 陈振华, 王慧敏, 等. 镁合金的腐蚀与防护研究进展[J]. 机械工程材料, 2005, 29(5): 1-4. CHENG Y L, CHEN Z H, WANG H M, et al. Progress in the research of corrosion and protection of magnesium alloys[J]. Materials for Mechanical Engineering, 2005, 29(5): 1-4. |
| [4] |
PHUONG N V, GUPTA M, MOON S. Enhanced corrosion performance of magnesium phosphate conversion coating on AZ31 magnesium alloy[J]. Transactions of Nonferrous Metals Society of China, 2017, 27: 1087-1095. DOI:10.1016/S1003-6326(17)60127-4 |
| [5] |
GUSIEVA K, DAVIES C H J, SCULLY J R, et al. Corrosion of magnesium alloys:the role of alloying[J]. International Materials Reviews, 2015, 60: 169-194. DOI:10.1179/1743280414Y.0000000046 |
| [6] |
PERRAULT G G. The potential-pH diagram of the magnesium-water system[J]. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 1974, 51(1): 107-119. DOI:10.1016/S0022-0728(74)80298-6 |
| [7] |
王新印, 周亚茹, 张鉴清, 等. 镁及其合金腐蚀析氢的研究现状[J]. 装备环境工程, 2015, 12(4): 129-141. WANG X Y, ZHOU Y R, ZHANG J Q, et al. Research status of hydrogen evolution during magnesium alloy's corrosion process[J]. Equipment Environment Engineering, 2015, 12(4): 129-141. |
| [8] |
曹楚南. 腐蚀电化学原理[M]. 第3版.北京: 化学工业出版社, 2008. CAO C N. Principles of electrochemistry of corrosion[M]. 3rd ed. Beijing: Chemical Industry Press, 2008. |
| [9] |
CHEN J, DONG J H, WANG J Q, et al. Effect of magnesium hydride on the corrosion behavior of an AZ91 magnesium alloy in sodium chloride solution[J]. Corrosion Science, 2008, 50(12): 3610-3613. DOI:10.1016/j.corsci.2008.09.013 |
| [10] |
李瑛, 宋光玲, 林海潮, 等. 金属镁在腐蚀介质中界面结构特征与负差数效应关系研究[J]. 腐蚀科学与防护技术, 1999, 11(4): 202-208. LI Y, SONG G L, LIN H C, et al. Study on the relationship between the corrosion interface structure and negative difference effect for pure magnesium[J]. Corrosion Science and Protection Technology, 1999, 11(4): 202-208. |
| [11] |
SONG G, ATRENS A, STJOHN D, et al. The electrochemical corrosion of pure magnesium in 1 N NaCl[J]. Corrosion Science, 1997, 39: 855-875. DOI:10.1016/S0010-938X(96)00172-2 |
| [12] |
THOMAS S, MEDHEKAR N V, FRANKEL G S, et al. Corrosion mechanism and hydrogen evolution on Mg[J]. Current Opinion in Solid State and Materials Science, 2015, 19: 85-94. DOI:10.1016/j.cossms.2014.09.005 |
| [13] |
BEETZ W. On the development of hydrogen from the anode[J]. Philosophical Magazine and Journal of Science, 1866, 216(32): 269-278. |
| [14] |
ESMAILY M, SVENSSON J E, FAJARDO S, et al. Fundamentals and advances in magnesium alloy corrosion[J]. Progress in Materials Science, 2017, 89: 92-193. DOI:10.1016/j.pmatsci.2017.04.011 |
| [15] |
SACEANT J M, TESSIER D. Convolution potential sweep voltammetry V. Determination of charge transfer kinetics deviating from the Butler-Volmer behaviour[J]. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 1975, 65(1): 57-66. DOI:10.1016/0368-1874(75)85105-7 |
| [16] |
CURIONI M. The behaviour of magnesium during free corrosion and potentiodynamic polarization investigated by real-time hydrogen measurement and optical imaging[J]. Electrochimica Acta, 2014, 120: 284-292. DOI:10.1016/j.electacta.2013.12.109 |
| [17] |
CURIONI M, SALAMONE L, SCENINI F, et al. A mathematical description accounting for the superfluous hydrogen evolution and the inductive behaviour observed during electrochemical measurements on magnesium[J]. Electrochimica Acta, 2018, 274: 343-352. DOI:10.1016/j.electacta.2018.04.116 |
| [18] |
SONG G L, ATRENS A. Corrosion mechanisms of magnesium alloys[J]. Advanced Engineering Materials, 1999, 1: 11-33. DOI:10.1002/(SICI)1527-2648(199909)1:1<11::AID-ADEM11>3.0.CO;2-N |
| [19] |
CURIONI M, SCENINI F, MONETTA T, et al. Correlation between electrochemical impedance measurements and corrosion rate of magnesium investigated by real-time hydrogen measurement and optical imaging[J]. Electrochimica Acta, 2015, 166: 372-384. DOI:10.1016/j.electacta.2015.03.050 |
| [20] |
WILLIAMS G, BIRBILIS N, MCMURRAY H N. The source of hydrogen evolved from a magnesium anode[J]. Electrochemistry Communications, 2013, 36: 1-5. DOI:10.1016/j.elecom.2013.08.023 |
| [21] |
FAJARDO S, FRANKEL G S. Effect of impurities on the enhanced catalytic activity for hydrogen evolution in high purity magnesium[J]. Electrochimica Acta, 2015, 165: 255-267. DOI:10.1016/j.electacta.2015.03.021 |
| [22] |
SALLEH S H, THOMAS S, YUWONO J A, et al. Enhanced hydrogen evolution on Mg (OH)2 covered Mg surfaces[J]. Electrochimica Acta, 2015, 161: 144-152. DOI:10.1016/j.electacta.2015.02.079 |
| [23] |
TAHERI M, KISH J R, BIRBILIS N, et al. Towards a physical description for the origin of enhanced catalytic activity of corroding magnesium surfaces[J]. Electrochimica Acta, 2014, 116: 396-403. DOI:10.1016/j.electacta.2013.11.086 |
| [24] |
HOCHE D, BLAWERT C, LAMAKA S V, et al. The effect of iron re-deposition on the corrosion of impurity-containing magnesium[J]. Physical Chemistry Chemical Physics, 2016, 18: 1279-1291. DOI:10.1039/C5CP05577F |
| [25] |
LAMAKA S V, HOCHE D, PETRAUSKAS R P, et al. A new concept for corrosion inhibition of magnesium:suppression of iron re-deposition[J]. Electrochemistry Communications, 2016, 62: 5-8. DOI:10.1016/j.elecom.2015.10.023 |
| [26] |
STRAUMANIS M E, BHATIA B K. Disintegration of magnesium while dissolving anodically in neutral and acidic solutions[J]. Journal of the Electrochemical Society, 1963, 110(5): 357-360. DOI:10.1149/1.2425763 |
| [27] |
CURIONI M, TORRESCANO-ALVAREZ J M, YANG Y F, et al. Application of side-view imaging and real-time hydrogen measurement to the investigation of magnesium corrosion[J]. Corrosion, 2017, 73: 463-470. DOI:10.5006/2299 |
| [28] |
FRANKEL G S, FAJARDO S, LYNCH B M. Introductory lecture on corrosion chemistry:a focus on anodic hydrogen evolution on Al and Mg[J]. Faraday Discussions, 2015, 180: 11-33. DOI:10.1039/C5FD00066A |
| [29] |
TAYLOR C D. A first-principles surface reaction kinetic model for hydrogen evolution under cathodic and anodic conditions on magnesium[J]. Journal of the Electrochemical Society, 2016, 163(9): 602-608. DOI:10.1149/2.1171609jes |
| [30] |
GOMBERG M. The reducing effect of the binary system (MgX2+Mg) upon organic compounds in anhydrous solvents[J]. Recueil des Travaux Chimiques des Pays-bas, 2010, 48: 847-851. DOI:10.1002/recl.19290480815 |
| [31] |
PETTY R L, DAVIDSON A W, KLEINBERG J. The anodic oxidation of magnesium metal:evidence for the existence of unipositive magnesium[J]. Journal of the American Chemical Society, 1954, 76(2): 363-366. DOI:10.1021/ja01631a013 |
| [32] |
SAMANIEGO A, HURLEY B L, FRANKEL G S. On the evidence for univalent Mg[J]. Journal of Electroanalytical Chemistry, 2015, 737: 123-128. DOI:10.1016/j.jelechem.2014.04.013 |
| [33] |
ATRENS A, SONG G L, LIU M, et al. Review of recent developments in the field of magnesium corrosion[J]. Advanced Engineering Materials, 2015, 17(4): 400-453. DOI:10.1002/adem.201400434 |
| [34] |
ROSSRUCKER L, MAYRHOFER K J J, FRANKEL G S, et al. Investigating the real time dissolution of Mg using online analysis by ICP-MS[J]. Journal of The Electrochemical Society, 2014, 161(3): 115-119. |
| [35] |
SWIATOWSKA J, VOLOVITCH P, OGLE K. The anodic dissolution of Mg in NaCl and Na2SO4 electrolytes by atomic emission spectroelectrochemistry[J]. Corrosion Science, 2010, 52(7): 2372-2378. DOI:10.1016/j.corsci.2010.02.038 |
| [36] |
WILLIAMS G, MCMURRAY H N. Localized corrosion of magnesium in chloride containing electrolyte studied by a scanning vibrating electrode technique[J]. Journal of the Electrochemical Society, 2008, 155(7): 340-349. DOI:10.1149/1.2918900 |
| [37] |
BIRBILIS N, KING A D, THOMAS S. Evidence for enhanced catalytic activity of magnesium arising from anodic dissolution[J]. Electrochimica Acta, 2014, 132: 277-283. DOI:10.1016/j.electacta.2014.03.133 |
| [38] |
FRANKEL G S, SAMANIEGO A, BIRBILIS N. Evolution of hydrogen at dissolving magnesium surfaces[J]. Corrosion Science, 2013, 70: 104-111. DOI:10.1016/j.corsci.2013.01.017 |
| [39] |
GORE P, CAIN T W, LAIRD J, et al. Enrichment efficiency of noble alloying elements on magnesium and effect on hydrogen evolution[J]. Corrosion Science, 2019, 151: 206-218. DOI:10.1016/j.corsci.2019.02.026 |
| [40] |
MARSH G A, SCHASCHL E. The difference effect and the chunk effect[J]. Journal of the Electrochemical Society, 1960, 107(12): 960-965. DOI:10.1149/1.2427579 |
| [41] |
HOEY G R, COHEN M. Corrosion of anodically and cathodically polarized magnesium in aqueous media[J]. Journal of the Electrochemical Society, 1958, 105(5): 245-250. DOI:10.1149/1.2428817 |
| [42] |
UNOCIC K A, ELSENTRIECY H H, BRADY M P, et al. Transmission electron microscopy study of aqueous film formation and evolution on magnesium alloys[J]. Journal of the Electrochemical Society, 2014, 161(6): 302-311. DOI:10.1149/2.024406jes |
| [43] |
DANAIE M, ASMUSSEN R M, JAKUPI P. The role of aluminum distribution on the local corrosion resistance of the microstructure in a sand-cast AM50 alloy[J]. Corrosion Science, 2013, 77: 151-163. DOI:10.1016/j.corsci.2013.07.038 |
| [44] |
ASMUSSEN R M, JAKUPI P, DANAIE M. Tracking the corrosion of magnesium sand cast AM50 alloy in chloride environments[J]. Corrosion Science, 2013, 75: 114-122. DOI:10.1016/j.corsci.2013.05.022 |
| [45] |
PERRAULT G G. Potentiostatic study of the magnesium electrode in aqueous solution[J]. Journal of Electroanalytical Chemistry, 1970, 27(1): 47-58. |
| [46] |
GULBRANDSEN E. Anodic behaviour of Mg in HCO3-/CO32- buffer solutions. Quasi-steady measurements[J]. Electrochimica Acta, 1992, 37(8): 1403-1412. DOI:10.1016/0013-4686(92)87014-Q |
| [47] |
BENDER S, GOELLNER J, HEYN A, et al. A new theory for the negative difference effect in magnesium corrosion[J]. Materials and Corrosion, 2012, 63(8): 707-712. DOI:10.1002/maco.201106225 |
| [48] |
BIRBILIS N, CAIN T, LAIRD J S, et al. Nuclear microprobe analysis for determination of element enrichment following magnesium dissolution[J]. ECS Electrochemistry Letters, 2015, 4: 34-37. DOI:10.1149/2.0081510eel |
| [49] |
FAJARDO S, GLOVER C F, WILLIAMS G, et al. The source of anodic hydrogen evolution on ultra high purity magnesium[J]. Electrochimica Acta, 2016, 212: 510-521. DOI:10.1016/j.electacta.2016.07.018 |
| [50] |
FAJARDO S, BOSCH J, FRANKEL G S, et al. Anomalous hydrogen evolution on AZ31, AZ61 and AZ91 magnesium alloys in unbuffered sodium chloride solution[J]. Corrosion Science, 2019, 146: 163-171. DOI:10.1016/j.corsci.2018.10.039 |
| [51] |
FAJARDO S, GHARI O, BIRBILIS N, et al. Investigating the effect of ferrous ions on the anomalous hydrogen evolution on magnesium in acidic ferrous chloride solution[J]. Journal of the Electrochemical Society, 2018, 165(13): 916-925. DOI:10.1149/2.0951813jes |
| [52] |
CHRISTOFER L, WALLINDER I O, TIDBLAD J, et al. Atmospheric corrosion[M]. 2nd ed. New Jersey: John Wiley & Sons Inc, 2016.
|
| [53] |
LIU H G, CAO F Y, SONG G L, et al. Review of the atmospheric corrosion of magnesium alloys[J]. Journal of Materials Science & Technology, 2019, 35: 2003-2016. |
| [54] |
SURENDRALAL S, TODOROVA M, FINNIS M W, et al. First-principles approach to model electrochemical reactions:understanding the fundamental mechanisms behind Mg corrosion[J]. Physical Review Letter, 2018, 120: 246801. DOI:10.1103/PhysRevLett.120.246801 |
| [55] |
YUWONO J A, TAYLOR C D, FRANKEL G S, et al. Understanding the enhanced rates of hydrogen evolution on dissolving magnesium[J]. Electrochemistry Communications, 2019, 104: 106482. DOI:10.1016/j.elecom.2019.106482 |
| [56] |
YUWONO J A, BIRBILIS N, TAYLOR C D, et al. Aqueous electrochemistry of the magnesium surface:thermodynamic and kinetic profiles[J]. Corrosion Science, 2019, 147: 53-68. DOI:10.1016/j.corsci.2018.10.014 |
| [57] |
THOMAS S, GHARI O, SALLEH S H, et al. On the effect of Fe concentration on Mg dissolution and activation studied using atomic emission spectroelectrochemistry and scanning electrochemical microscopy[J]. Electrochimica Acta, 2016, 210: 271-284. DOI:10.1016/j.electacta.2016.05.164 |
| [58] |
SALLEH S H, BIRBILIS N, MUSAMEH M, et al. On the development and application of an in-house fabricated Mg2+ion selective microelectrode (ISME) for assessing Mg corrosion[J]. Journal of the Electrochemical Society, 2018, 165(11): 771-776. DOI:10.1149/2.0591811jes |