 2020, Vol. 48
2020, Vol. 48文章信息
- 李英民, 马鸣檀, 任玉艳, 刘桐宇
- LI Ying-min, MA Ming-tan, REN Yu-yan, LIU Tong-yu
- 稀土La掺杂Mg2Si的几何结构、弹性性能和电子结构的第一性原理研究
- First principles study on geometry structure, elastic property and electronic structure of La-doped Mg2Si
- 材料工程, 2020, 48(4): 100-107
- Journal of Materials Engineering, 2020, 48(4): 100-107.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2019.000153
-
文章历史
- 收稿日期: 2019-02-25
- 修订日期: 2019-08-28




2. 潍坊科技学院 机械工程学院, 山东 潍坊 262700
2. School of Mechanical Engineering, Weifang University of Science and Technology, Weifang 262700, Shandong, China
Mg2Si作为一种金属间化合物,具有密度低、熔点高、弹性模量高、抗腐蚀和抗氧化性能好的优异特点,现如今其作为一种轻质热电材料被广泛应用[1]。但是单相Mg2Si处于室温到450 ℃的温度范围内则会出现本征脆性,室温下几乎没有延展性。而且在传统铸造条件下,形成的初生Mg2Si相常呈现为树枝状/尖角易撕裂基体且晶粒较为粗大,会降低基体材料的性能[2-3]。因此,必须提高Mg2Si的韧性以满足生产实际的需求。掺杂是改变材料力学性能的有效方法之一。
掺杂稀土元素可以有效地改变材料的微观结构,影响材料的宏观性能[4]。稀土钪(Sc)对Mg2Si/Mg-Al复合材料有显著的变质效果[5],而适量的Sc使得A357铸造合金的晶粒尺寸明显减小,强度和延展性得到了提高[6]。李英民等[7]将不同含量的稀土元素镨(Pr)加入Al-30%(质量分数,下同)Mg2Si中,当Pr的加入量为0.6%~0.8%时,Mg2Si初生相得到明显细化,形状规则且分布最为均匀。稀土铈(Ce)也能改变初生Mg2Si相的形貌[8-9]。Chen等[10]也利用第一性原理的方法,计算了稀土钇(Y)掺杂SrSi2的介电函数和吸收谱,表明其是一种有前途的介电材料。稀土Y可以改变初生相Mg2Si的晶粒尺寸,当加入Y的量为0.6%时,Al-30% Mg2Si复合材料晶粒的细化效果最好,力学性能提高最为显著[11]。稀土La[12]可以使Mg2Si颗粒变得更加细小, Al11La3相可阻止Mg2Si相长大且铸态Mg2Si/Al基复合材料的力学性能得到改善。刘政等[13]还从理论上研究了稀土Y的细化机制,通过计算的方法,发现Y可以作为初生相Mg2Si的异质形核质点从而细化颗粒,同时Y与Al相互作用形成Al3Y相可阻止Mg2Si相长大。崔策等[14]发现往Mg2Si/AZ91复合材料中添加适量的稀土铒(Er)不仅可以改变基体和Mg2Si初生相的晶粒尺寸,还改变了Mg2Si初生相的形貌与分布,合金的抗拉强度和伸长率得到了明显的提升。柏世梅等[15]从实验方面研究了稀土元素Er,Ce复合作用下对初生Mg2Si相的改性机制。李晓燕等[16]发现稀土Er的加入对A356铝合金铸态组织中的共晶硅起到了变质作用。刘文祎等[17]发现稀土钆(Gd)可以有效地对共晶硅进行细化,但是对其形貌影响不大。因此,现如今Mg2Si掺杂稀土元素进行改性已经成为该材料的研究热点,但是,在关于稀土掺杂改性的理论研究方面尚不完善。本工作采用第一性原理计算,对稀土La掺杂Mg2Si的几何结构、弹性性能和电子结构进行了计算,期望能为稀土掺杂Mg2Si改性提供理论依据。
1 理论模型和计算方法 1.1 计算模型Mg2Si为几何密排相,其晶体结构为面心立方,空间点群为Fm3m,具有反萤石结构,属于正常价金属间化合物。其每个晶胞内有8个镁原子和4个硅原子,晶格常数为a=b=c=0.635 nm[18],α=β=γ=90°,其中镁的原子坐标为(0.25,0.25,0.25),硅的原子坐标为(0,0,0),晶胞结构如图 1(a)所示。稀土镧掺杂后的晶胞模型如图 1(b)~(d)所示。计算时镧元素的占位情况分别为间隙到Mg2Si晶胞中(图 1(b))、置换晶胞中Mg原子的位置(图 1(c))和置换晶胞中Si原子的位置(图 1(d))。Mg原子和Si原子各个位置都等效,掺杂位置可确定。间隙固溶体Mg8Si4La占位情况多样,但由于掺杂量少,实验不易确定位置,因此,本工作选择被8个Mg原子包围的体心位置。

|
图 1 晶体结构 (a)Mg2Si; (b)Mg8Si4La; (c)Mg7Si4La; (d)Mg8Si3La Fig. 1 Crystal structure (a)Mg2Si; (b)Mg8Si4La; (c)Mg7Si4La; (d)Mg8Si3La |
本工作计算采用基于密度泛函理论(DFT)的CASTEP(Cambridge serial total energy package)软件包[19]进行的。首先利用Materials Studio软件[20]建立晶胞模型,其次利用BFGS算法[21]对掺杂前后的晶胞进行几何优化,得到稳定的体系结构,再对体系的性质进行计算。本工作将截断能Ecut设置为340.0 eV,电子之间相互作用的交换和相关势的函数选择广义梯度近似(generalized gradient approximation, GGA)[22]中的PBE进行校正,势函数选用倒易空间中表述的超软(Ultrasoft)赝势[23]。布里渊区积分采用Monkhors-Pack[24]形式的高对称k点方法,k点网格设置4×4×4。计算过程中最大的能量变化收敛值为1×10-5 eV/atom,最大应力收敛参数为0.05 GPa。
2 结果与分析 2.1 平衡晶格常数表 1为Mg2Si晶胞掺杂稀土元素La前后优化的晶格常数,可以看出Mg8Si4晶格常数的计算值和实验值误差很小,误差值为0.55%,这表明第一性原理计算结果的合理性和有效性。由表 1还可以发现,当稀土La元素以不同方式固溶在Mg2Si晶胞中时,首先掺杂后的晶格常数和晶胞体积都有所增大,其次由于固溶的位置不同且稀土元素La的原子半径(0.187 nm)大于Mg的原子半径(0.160 nm)和Si的原子半径(0.134 nm),因此导致了其掺杂后的平衡晶格常数的值也不一样。这表明利用稀土元素La掺杂可以改变Mg2Si的结构参数。
| Sample | a/nm | b/nm | c/nm | V/(10-3nm3) |
| Mg2Si (experimental) | 0.6350 | 0.6350 | 0.6350 | 256.048 |
| Mg2Si (calculated) | 0.6385 | 0.6385 | 0.6385 | 260.305 |
| Mg8Si4La | 0.6652 | 0.6652 | 0.6652 | 294.345 |
| Mg7Si4La | 0.6573 | 0.6573 | 0.6573 | 283.982 |
| Mg8Si3La | 0.6758 | 0.6726 | 0.6758 | 307.182 |
由于第一性原理计算默认压力是恒定的,因此,晶胞体积不受压力影响,此时形成焓与反应热相同。故形成焓是物质反应后释放或吸收的热量,其大小可以突出晶体结构是否稳定,当反应热为负值时,其值越小,此晶体结构的稳定性越好。结合能是几个粒子从自由状态结合成为一个复合粒子时所释放的能量。结合能的绝对值越大,分子(原子或原子核)的结构就越稳定。形成焓(Hform)和结合能(Ecoh)计算公式[25]如下:
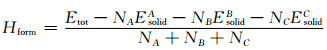
|
(1) |

|
(2) |
式中:NA,NB,NC表示在各晶胞结构模型中的原子个数;Etot代表了各晶胞的总能量;EsolidA,EsolidB, EsolidC分别表示固体原子A,B,C单个原子的能量。而EatomA,EatomB,EatomC则分别代表了A,B,C原子在晶胞点阵中的能量,计算结果如表 2、表 3所示。
| Sample | Etot/eV | Hform/(kJ·mol-1) | Ecoh/(kJ·mol-1) |
| Mg2Si | -8222.6772 | -19.31 | -311.52 |
| Mg8Si4La | -9083.9765 | -8.06 | -314.36 |
| Mg7Si4La | -8110.2477 | -10.56 | -327.82 |
| Mg8Si3La | -8970.5835 | 3.56 | -245.13 |
表 2给出了本工作中的Mg, Si和La的Esolid与Eatom的计算结果,并将本工作的计算结果与其他研究人员的结果进行了对比,对比结果表明误差在5%之内,证明了本工作计算结果的可靠性。综合表 2和表 3,首先在形成焓方面,由于形成焓的值为负值时结构才会稳定存在,而Mg8Si3La的形成焓的值为正值,因此稀土La元素置换Si位置形成的结构不能稳定存在;其次,Mg8Si4La和Mg7Si4La的形成焓均为负值,说明两种结构是可以形成的,但Mg7Si4La形成焓的值更小,表明比起形成间隙固溶体,稀土元素La更易占据Mg2Si中的Mg位置,形成置换固溶体,但是Mg7Si4La的形成焓大于Mg2Si的形成焓,所以说明Mg2Si是更稳的存在。最后在结合能方面,Mg7Si4La结合能的绝对值大于Mg8Si4La,也说明了稀土元素置换Mg位置的间隙固溶体更易优先形成。
2.3 弹性性能弹性常数是表征材料弹性的量,对于不同的单晶体来说,弹性常数的数量也不相同,而且体系的对称性对弹性常数的数量起到一定的决定性作用。例如,任何方向测量性质都相同的各向同性介质只有两个独立的弹性常数;单对称材料有13个独立常数;由于立方晶系的对称性,导致本次工作的各晶胞只存在3个独立常数,分别为C11, C12和C44,Mg2Si及其固溶体的弹性常数如表 4所示。
| Sample | Elastic constant/GPa | C11-C12 | C12-C44 | ||
| C11 | C12 | C44 | |||
| Mg2Si | 114.328 | 23.301 | 45.253 | 91.027 | -21.952 |
| Mg2Si[28] | 126 | 26 | 48.5 | 100 | -22.5 |
| Mg2Si[29] | 114.50 | 21.50 | 45.60 | 93.00 | -24.10 |
| Mg8Si4La | 1.420 | 67.149 | 19.260 | -65.729 | 47.889 |
| Mg7Si4La | 95.192 | 26.976 | 24.080 | 68.216 | 2.896 |
| Mg8Si3La | 20.545 | 37.478 | 6.953 | -16.933 | 30.525 |
首先由Born力学稳定条件[30]可知,弹性常数可以判断结构的稳定性,具体公式如下:
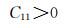
|
(3) |
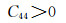
|
(4) |
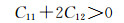
|
(5) |

|
(6) |
由表 4得知Mg8Si4La和Mg8Si3La均不满足Born力学稳定条件,证明这两种结构均不能稳定存在,而且此时的间隙固溶体是由La原子间隙在Mg2Si晶胞的体心位置形成的,根据晶体学理论,晶体体心处的间隙最大,相对其余间隙位置更易形成间隙固溶体,而此计算中Mg2Si晶胞在体心位置掺杂La原子后形成的Mg8Si4La恰恰不能稳定存在,因此表明了其他位置的间隙固溶体也很难稳定存在,所以将Mg7Si4La和Mg8Si4La和Mg8Si3La这3种结构进行比较,证明了置换固溶体Mg7Si4La是稀土镧掺杂后形成的固溶体中更稳定的存在。其次,这些弹性常数还可以预测出多晶的许多弹性性质,晶体的体模量(B),剪切模量(G),杨氏模量(E),泊松比(ν),以及各向异性系数(A)的计算公式如下[31]:
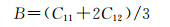
|
(7) |
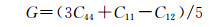
|
(8) |
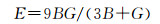
|
(9) |
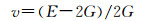
|
(10) |
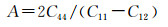
|
(11) |
文献[32]指出通过计算体模量与剪切模量的比值可以判断材料的塑性或脆性,即当B/G的比值小于1.75时,材料表现为脆性,比值越小,脆性越强;若比值大于1.75则材料表现为塑性,比值越大塑性也越强。从表 5中可以发现Mg2Si的B与G的比值小于1.75,说明Mg2Si为脆性,这与本征Mg2Si的性质相符,证明了计算的有效性;Mg7Si4La的B与G的比值大于1.75,说明此间隙固溶体呈现塑性。对于衡量材料的韧脆性问题,还可以用C12-C44[33]来判断,其值若小于0,则呈现脆性;反之为塑性。从表 4可以知道Mg2Si的C12-C44值为负,显脆性;Mg7Si4La的C12-C44值为正,显塑性。泊松比也可以用来表征材料的塑性[34],其值越大材料的相对塑性也越好,Mg2Si泊松比的值小于Mg7Si4La。综合以上3项指标可以发现稀土掺杂形成的置换固溶体能够在一定程度上降低Mg2Si的脆性。
杨氏模量E是表征材料刚度的重要参数,其值越大,刚度越大,材料塑性越差[35]。表 5的结果表明Mg7Si4La的塑性要高于Mg2Si的塑性。材料的弹性各向异性系数A表明材料出现微裂纹的可能性[33],A的值表示材料各向异性的程度,当材料为各向同性时,A=1。表 5所示的Mg7Si4La的A值与1较为接近,表明其偏离各向同性的程度较小,因此其出现微裂纹的可能性较小。
2.4 电子结构 2.4.1 态密度态密度是材料电子结构的重要表现形式之一,电子结构中电子的成键及相互作用情况是决定材料的稳定性的重要参数。图 2(a),(b)分别为Mg2Si和Mg7Si4La的总(分)态密度曲线图,蓝色虚线表示费米能级,对应能量值为0。本研究中各元素的价态电子为:Mg 2p63s2,Si 3s23p2和La 5s25p65d16s2。

|
图 2 Mg2Si(a)和Mg7Si4La(b)总(分)态密度图 Fig. 2 Total(partial) density of state of Mg2Si(a) and Mg7Si4La(b) |
由图 2知Mg2Si和Mg7Si4La的成键电子主要分布在-10~5 eV区间,在-10~-5 eV范围内,Mg2Si和Mg7Si4La的成键电子主要是由Si3s贡献的;在-5~0 eV范围内,Mg2Si的Si3p,Mg2p,Mg3s的轨道出现了明显的杂化现象,说明Mg和Si之间存在成键,Si3p对价带的贡献大于Mg原子,电子分布在Si原子周围,另外在0~5 eV范围内,Mg2Si成键电子主要是由Mg2p贡献的。对于Mg7Si4La,在-5~0 eV范围内,Si3p,Mg2p,Mg3s和La5d产生了轨道杂化,说明了Si,Mg和La之间存在成键效应;在0~5 eV范围内,Mg7Si4La的成键电子主要由Mg2p,Mg3s和La5d贡献;其次,根据赝能隙理论,在费米能级两侧分别有两个尖峰,而两个尖峰之间的DOS并不为零,两个峰之间的距离即赝能隙,其直接反映了该体系成键的共价性的强弱:越宽,说明共价性越强,所以Mg2Si的赝能隙要明显大于Mg7Si4La,即共价性更强。从图 2中还可以发现,Mg2Si费米能级对应的电子态密度为1.65 eV,经过La掺杂后的费米面向高能级区域偏移,使得费米能级对应的电子态密度增大,Mg7Si4La的费米能级对应的态密度为3.33 eV,说明掺杂后Mg7Si4La的导电性要高于Mg2Si的导电性。
2.4.2 Mulliken布居数研究晶体的Mulliken布居数对分析晶体结构稳定性的物理本质也有帮助[36]。Mulliken布居数也叫电子占据数,它可以判断体系中不同原子间的电荷转移情况和比较不同体系中离子键的强弱程度,而离子键的强弱程度又与电荷转移数有关,即每个原子转移平均电荷数越多,离子键就越强。在Mg2Si晶体结构中,由表 6可知,Si得到电子,电荷由Mg原子向Si原子转移,电荷转移总数约为5.36,每个Mg原子向Si原子转移电荷数量为0.67,体系中平均原子电荷数约为0.45。对于Mg7Si4La而言,电荷由La原子和Mg原子向Si原子转移,其中Mg原子分为三类,Si原子分为两类,电荷转移总数约为4.47,总转移电荷数除以体系中的12个有效原子,得到平均原子转移电荷数约为0.37。因此,二元相Mg2Si和三元相Mg7Si4La相比,两者皆具有一定的离子键特性。
| Sample | Atom | Number | Population | Transfer charge | Total transfer charge | ||
| s | p | d | |||||
| Mg2Si | Mg | 8 | 0.68 | 6.66 | - | 0.67 | 7.33 |
| Si | 4 | 1.63 | 3.70 | - | -1.33 | 5.36 | |
| Mg7Si4La | Mg(Ⅰ) | 3 | 0.76 | 6.70 | - | 0.55 | 7.45 |
| Mg(Ⅱ) | 3 | 0.79 | 6.73 | - | 0.48 | 7.52 | |
| Mg(Ⅲ) | 1 | 0.68 | 6.69 | - | 0.62 | 7.38 | |
| Si(Ⅰ) | 3 | 1.62 | 3.50 | - | -1.12 | 5.12 | |
| Si(Ⅱ) | 1 | 1.62 | 3.48 | - | -1.10 | 5.10 | |
| La | 1 | 1.82 | 6.10 | 2.33 | 0.76 | 10.24 | |
差分电荷密度是指当原子堆积成晶体以后,其构成的体系中电荷的重新分布。能够直观地反映电荷分布状态及键合能力。本工作分别截取了Mg2Si和Mg7Si4La的(110)面,对其进行差分电荷密度分析,结果如图 3(a),(b)所示。为了更加直观地观察到差分电荷密度图的效果,Mg2Si和Mg7Si4La的(110)面的等高线分别选取-0.05184 eV/nm到0.05184 eV/nm和-0.05212 eV/nm到0.05212 eV/nm之间。图 3(a)可以发现,在Mg2Si中,大部分电荷分布由Mg提供给Si原子,均匀分布在Si原子周围。因此,Mg—Mg之间是金属键;Mg—Si之间分布的电荷大量趋向硅原子,形成方向性较强的共价键,并伴有离子键特性;Si—Si之间的电子分布存在方向性,且方向相同,故存在一定的共价性。由图 3(b)可知,Mg—Mg金属键,Mg—Si共价键和Si—Si共价键也存在于Mg7Si4La中,另外,一些La原子将电子转移到Si原子,并且电子云的高密度区域倾向于Si周围的局部区域,形成La—Si离子键,说明Mg7Si4La是可以稳定存在的。

|
图 3 Mg2Si(a)和Mg7Si4La(b)的差分电荷密度 Fig. 3 Electronic charge density difference contour plots of Mg2Si(a) and Mg7Si4La(b) |
(1) 根据Born力学稳定性和形成焓的标准,Mg2Si和Mg7Si4La均可以稳定地存在于体系中,Mg8Si3La和Mg8Si4La均不能稳定存在,La掺杂的Mg2Si优先占据体系Mg原子的位置。
(2) 剪切模量、体模量、杨氏模量和泊松比的计算结果表明Mg2Si为脆性相,而Mg7Si4La为韧性相。稀土La掺杂形成的置换固溶体能够在一定程度上降低Mg2Si的脆性。
(3) 电子结构的计算结果表明,掺杂稀土La后,费米能级进入导带,提高了Mg2Si的导电性,Mg2Si和Mg7Si4La都带有一定的离子键特性;Mg2Si和Mg7Si4La的成键本质是Mg—Mg金属键、Si—Si共价键、Mg—Si共价键和La—Si离子键的结合。
| [1] |
FARAHANY S, GHANDVAR H, NORDIN N A, et al. Effect of primary and eutectic Mg2Si crystal modifications on the mechanical properties and sliding wear behaviour of an Al-20Mg2Si-2Cu-xBi composite[J]. Journal of Materials Science & Technology, 2016(11): 1083-1097. |
| [2] |
CHEN K, LI Z. Effect of comodification by Ba and Sb on the microstructure of Mg2Si/Mg-Zn3Si composite and mechanism[J]. Journal of Alloys and Compounds, 2014, 592: 196-201. DOI:10.1016/j.jallcom.2013.12.041 |
| [3] |
臧树俊, 周琦, 马勤, 等. 金属间化合物Mg2Si研究进展[J]. 铸造技术, 2006, 27(8): 866-870. ZANG S J, ZHOU Q, MA Q, et al. Research progress of intermetallic compound Mg2Si[J]. Foundry Technology, 2006, 27(8): 866-870. DOI:10.3969/j.issn.1000-8365.2006.08.027 |
| [4] |
屈敏, 刘鑫, 崔岩, 等. 稀土元素对原位合成TiB2/Al复合材料组织和性能的影响[J]. 材料工程, 2018, 46(3): 98-104. QU M, LIU X, CUI Y, et al. Effect of rare earth element on Microstructure and properties of in situ synthesized TiB2/Al composites[J]. Journal of Materials Engineering, 2018, 46(3): 98-104. |
| [5] |
林继兴, 高光亮, 牛丽媛, 等. Ca、Sc对原位Mg2Si/Mg-Al复合材料组织的影响[J]. 热加工工艺, 2010, 39(24): 122-123. LIN J X, GAO G L, NIU L Y, et al. Effects of Ca and Sc on the microstructure of in-situ Mg2Si/Mg-Al composites[J]. Hot Working Technology, 2010, 39(24): 122-123. DOI:10.3969/j.issn.1001-3814.2010.24.035 |
| [6] |
MUHAMMAD A, XU C, XUE J W, et al. High strength aluminum cast alloy:a Sc modification of a standard Al-Si-Mg cast alloy[J]. Materials Science and Engineering:A, 2014, 604: 122-126. DOI:10.1016/j.msea.2014.03.005 |
| [7] |
李英民, 董国升, 任玉艳, 等. 稀土镨对金属间化合物Mg2Si组织的影响[J]. 沈阳工业大学学报, 2010(3): 266-269. LI Y M, DONG G S, REN Y Y, et al. Effect of rare earth praseodymium on the microstructure of intermetallic compound Mg2Si[J]. Journal of Shenyang University of Technology, 2010(3): 266-269. |
| [8] |
杜军, 吕信裕, 李文芳. 稀土Ce对过共晶Mg-Si合金中初生Mg2Si相变质的影响[J]. 材料工程, 2011(6): 1-4. DU J, LU X Y, LI W F. Effect of rare earth Ce on primary Mg2Si phase change in overeutectic Mg-Si alloy[J]. Journal of Materials Engineering, 2011(6): 1-4. |
| [9] |
李克, 李健, 胡斐, 等. Ce对Mg-3Al-2.5Si合金中初生Mg2Si相的变质作用与机理[J]. 有色金属工程, 2019, 9(1): 23-28. LI K, LI J, HU F, et al. Metamorphism and mechanism of Ce on primary Mg2Si phase in Mg-3Al-2.5Si alloy[J]. Noferrous Metals Engineering, 2019, 9(1): 23-28. |
| [10] |
CHEN Z, YU M, CHEN T. First-principles study of the structural, electronic, and optical properties of Y-doped SrSi2[J]. Journal of Applied Physics, 2013, 113(4): 043515. DOI:10.1063/1.4788715 |
| [11] |
任玉艳, 刘桐宇, 李英民. 稀土元素钇对Al-30wt.%Mg2Si复合材料组织和力学性能的影响[J]. 复合材料学报, 2015, 32(5): 1367-1373. REN Y Y, LIU T Y, LI Y M. Effects of rare-earth element yttrium on microstructure and mechanical properties of Al-30wt.%Mg2Si composites[J]. Acta Materiae Compositae Sinica, 2015, 32(5): 1367-1373. |
| [12] |
刘政, 白光珠, 徐丽娜, 等. La对原位自生Mg2Si/Al复合材料组织及性能的影响[J]. 特种铸造及有色合金, 2015, 35(9): 897-901. LIU Z, BAI G Z, XU L N, et al. Effect of La on microstructure and properties of in situ autogenous Mg2Si/Al composites[J]. Special Casting & Non-ferrous Alloys, 2015, 35(9): 897-901. |
| [13] |
刘政, 白光珠, 罗浩林. Y对原位MgSi/Al基复合材料初生相Mg2Si的细化机制[J]. 有色金属科学与工程, 2016, 7(1): 28-33. LIU Z, BAI G Z, LUO H L. The fining mechanism of Y on primary phase Mg2Si of in-situ MgSi/Al matrix composites[J]. Nonferrous Metals Science and Engineering, 2016, 7(1): 28-33. |
| [14] |
崔策, 尧军平, 张磊, 等. Er对原位自生Mg2Si/AZ91复合材料组织和性能影响[J]. 特种铸造及有色合金, 2017(11): 82-85. CUI C, YAO J P, ZHANG L, et al. Effect of Er on microstructure and properties of in situ autogenous Mg2Si/AZ91 composites[J]. Special Casting & Nonferrous Alloys, 2017(11): 82-85. |
| [15] |
柏世梅, 尧军平, 刘辉, 等. 稀土Er和Ce复合改性对过共晶Mg-Si合金组织与性能的影响[J]. 复合材料学报, 2017, 34(6): 1300-1307. BAI S M, YAO J P, LIU H, et al. Effect of compound modification of rare earth Er and Ce on microstructure and properties of hypereutectic Mg-Si alloy[J]. Acta Materiae Compositae Sinica, 2017, 34(6): 1300-1307. |
| [16] |
李晓燕, 卢雅琳, 王健, 等. 稀土Er对A356铝合金微观组织和力学性能的影响[J]. 材料工程, 2018, 46(1): 67-73. LI X Y, LU Y L, WANG J, et al. Effect of rare earth erbium on microstructure and mechanical properties of A356 aluminum alloy[J]. Journal of Materials Engineering, 2018, 46(1): 67-73. |
| [17] |
刘文祎, 徐聪, 刘茂文, 等. 稀土元素Gd对Al-Si-Mg铸造合金微观组织和力学性能的影响[J]. 材料工程, 2019, 47(6): 129-135. LIU W Y, XU C, LIU M W, et al. Effect of rare earth element Gd on microstructure and mechanical properties of Al-Si-Mg cast alloy[J]. Journal of Materials Engineering, 2019, 47(6): 129-135. |
| [18] |
BOULET P, VERSTRAETE M J, CROCOMBETTE J P, et al. Electronic properties of the MgSi thermoelectric material investigated by linear-response density-functiona theory[J]. Computational Materials Science, 2011, 50(3): 847-851. DOI:10.1016/j.commatsci.2010.10.020 |
| [19] |
黄志伟, 赵宇宏, 侯华, 等. V掺杂Ni3Al点缺陷结构及合金化效应的第一性原理研究[J]. 稀有金属材料与工程, 2011, 40(12): 2136-2141. HUANG Z W, ZHAO Y H, HOU H, et al. Study on the first-principles of V-doped Ni3Al point defect structure and alloying effect[J]. Rare Metal Materials and Engineering, 2011, 40(12): 2136-2141. |
| [20] |
FAN X H, XU B, XU Y, et al. Application of materials studio modeling in crystal structure[J]. Advanced Materials Research, 2013, 706/708(1): 7-10. |
| [21] |
FISCHER T H, ALMLOF J. General methods for geometry and wave function optimization[J]. Journal of Physical Chemistry, 1992, 96(24): 9768-9774. DOI:10.1021/j100203a036 |
| [22] |
PERDEW J P, YUE W. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Physical Review:B, 1992, 45(23): 13244-13249. DOI:10.1103/PhysRevB.45.13244 |
| [23] |
BOUCHAUD J P, ZERAH P G. The theory of ultrasoft magnetic films[J]. Journal of Applied Physics, 1990, 68(7): 3783-3785. DOI:10.1063/1.346305 |
| [24] |
WISESA P, MCGILL K A, MUELLER T. Efficient generation of generalized Monkhorst-Pack grids through the use of informatics[J]. Physical Review:B, 2016, 93(15): 155109. DOI:10.1103/PhysRevB.93.155109 |
| [25] |
傅利, 赵宇宏, 杨晓敏, 等. Mg-Al-Si-Ca合金系金属间化合物的电子结构和力学性能的第一性原理计算[J]. 稀有金属材料与工程, 2014, 43(11): 2733-2738. FU L, ZHAO Y H, YANG X M, et al. Primary principle calculation of the electronic structure and mechanical properties of Mg-Al-Si-Ca alloy series intermetallic compounds[J]. Rare Metal Materials and Engineering, 2014, 43(11): 2733-2738. |
| [26] |
张乐婷, 侯华, 文志勤, 等. Mg2Si和MgCu2相电子结构、弹性性能第一性原理研究[J]. 测试科学与仪器, 2017, 8(3): 289-294. ZHANG L T, HOU H, WEN Z Q, et al. Electronic structural, elastic properties of MgCu2 and Mg2Si phases based on first-principles calculations[J]. Journal of Measurement Science and Instrumentation, 2017, 8(3): 289-294. DOI:10.3969/j.issn.1674-8042.2017.03.013 |
| [27] |
FAN W H, CHEN R X, HAN P D, et al. First-principle study of electronic structures of Y-doped Mg2Si[J]. Materials Science Forum, 2011, 689: 102-107. DOI:10.4028/www.scientific.net/MSF.689.102 |
| [28] |
YU B H, LIU M L, CHEN D. First principles study of structural, electronic and elastic properties of Mg2Si polymorphs[J]. Acta Physica Sinica-Chinese Edition, 2011, 60(8): 298-300. |
| [29] |
刘娜娜. Mg2Si弹性性质及热力学性质的第一性原理计算[J]. 材料导报, 2009, 23(1): 278-280. LIU N N. Calculation of first principles of elastic and thermodynamic properties of Mg2Si[J]. Materials Review, 2009, 23(1): 278-280. |
| [30] |
BOULET P, VERSTRAETE M J, CROCOMBETTE J P, et al. Electronic properties of the Mg2Si thermoelectric material investigated by linear-response density-functional theory[J]. Computational Materials Science, 2011, 50(3): 847-851. DOI:10.1016/j.commatsci.2010.10.020 |
| [31] |
FU L, ZHAO Y, YANG L, et al. Electronic and elastic properties of Al4Ce binary compound under pressure via first-principles[J]. Superlattices & Microstructures, 2014, 69(5): 76-86. |
| [32] |
PUGH S F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals[J]. Philosophical Magazin, 1954, 45(367): 823-843. |
| [33] |
FU C L, WANG X, YE Y Y, et al. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation[J]. Intermetallics, 1999, 7(2): 179-184. DOI:10.1016/S0966-9795(98)00018-1 |
| [34] |
YU W Y, WANG N, XIAO X B, et al. First-principles investigation of the binary AB2 type laves phase in Mg-Al-Ca alloy:electronic structure and elastic properties[J]. Solid State Sciences, 2009, 11(8): 1400-1407. DOI:10.1016/j.solidstatesciences.2009.04.017 |
| [35] |
TVERGAARD V, HUTCHINSON J W. Microcracking in ceramics induced by thermal expansion or elastic anisotropy[J]. Journal of the American Ceramic Society, 1988, 71(3): 157-166. DOI:10.1111/j.1151-2916.1988.tb05022.x |
| [36] |
MAO P, BO Y U, LIU Z, et al. Mechanical properties and electronic structures of Mg-Cu, Mg-Ca and Mg-Zn-La phases by first principles calculations[J]. Transactions of Nonferrous Metals Society of China, 2014, 24(9): 2920-2929. DOI:10.1016/S1003-6326(14)63427-0 |