 2020, Vol. 48
2020, Vol. 48文章信息
- 刘继涛, 钏定泽, 杨泽斌, 陈希亮, 颜廷亭, 陈庆华
- LIU Ji-tao, CHUAN Ding-ze, YANG Ze-bin, CHEN Xi-liang, YAN Ting-ting, CHEN Qing-hua
- 氨基酸/羟基磷灰石复合材料的制备与表征及其在酸蚀牛牙釉质体外再矿化中的应用
- Synthesis and characterization of amino acids/hydroxyapatite composites for in vitro remi-neralization of acid-etched bovine enamel
- 材料工程, 2020, 48(2): 100-107
- Journal of Materials Engineering, 2020, 48(2): 100-107.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2019.000259
-
文章历史
- 收稿日期: 2019-03-21
- 修订日期: 2019-09-12




2. 云南白药集团股份有限公司, 昆明 650500
2. Yunnan Baiyao Group Co., Ltd., Kunming 650500, China
牙釉质是人体中最硬的矿化组织,含有约96%(质量分数,下同)的无机矿物质、1%的有机基质及少量的水[1]。牙釉质中的无机矿物主要是羟基磷灰石(HAP,Ca10(PO4)6(OH)2)晶体,具有六方形态。其特殊性质源于平行排列且垂直于牙釉质表面的纳米级HAP棱柱结构[2]。由于致龋菌产酸或酸性饮食的影响,牙釉质容易发生脱矿导致早期龋齿的形成。目前氟化物是常用的防龋方法,但其有导致氟斑牙的风险。近年来,酪蛋白磷酸肽稳定的无定形磷酸钙(CPP-ACP)常被用于早期龋齿再矿化。其他使用蛋白质或仿生大分子的再矿化方法也受到了极大的关注。如重组牙釉蛋白[3],模拟牙釉蛋白多肽[4-5]和PAMAM树枝状大分子[6]等。这些方法均促进了生物矿化并在牙釉质表面生成了HAP晶体。但由于这些蛋白质、多肽或仿生大分子的提纯或制备非常昂贵,限制了其进一步应用。同时,因大分子物质难以扩散至表面下脱矿区域,这类仿生矿化方法对牙釉质深层脱矿区域的修复作用有限。
HAP具有优异的生物相容性,已被广泛应用于组织工程领域[7]。另外,人工制备的HAP已被应用于酸蚀牙釉质的再矿化研究[8],其主要利用HAP纳米粒子的尺寸效应,在牙釉质表面空腔内形成不紧密的矿化层。或者利用HAP对细菌的吸附性能,干扰致龋菌和牙釉质表面之间的结合[9]。此类HAP材料的结晶度较高,因此其再矿化效果有限。
氨基酸(AA)是蛋白质的基本组成单位。在细胞介导的有机-无机生物矿化过程中,极性AA和带负电荷的AA发挥了重要的作用。已知釉质发生过程中的釉质基质均富含丝氨酸(Ser)、天冬氨酸(Asp)和谷氨酸(Glu)等残基。这类AA残基可能在生物矿化过程中参与了HAP形核和定向生长过程[10]。其中Ser残基在釉原蛋白中起重要作用,且与釉原蛋白和磷酸钙之间的相互作用有关[11]。Li等[12]研究发现,Glu可诱导具有20 nm左右晶粒尺寸的HAP颗粒叠加,在脱矿牙釉质表面上形成有序排列的釉质样结构。由于这种纳米HAP颗粒的表面能非常高,很容易团聚从而影响其再矿化效率。Wu等[13]研究发现,Asp和Glu可诱导脱矿牙釉质表面原位合成的碳酸钙,转化为有序排列的碳酸化HAP。但此方法需要在极端条件下通过两步法实现,难以实现体内应用。具有端羟基的Ser和端羧基的Asp与Glu均对HAP具有高亲和力,较小的分子尺寸也有利于其在脱矿牙釉质的离子通道中扩散。此外,当沿c轴取向时,HAP带正电[14]。因此,Ser,Asp和Glu的分子可以通过静电吸附与带正电的釉质棱镜强烈结合,并促进Ca2+和PO43-在表面下釉质龋的局部过饱和。同时有研究证实[15],与其他种类AA参与制备的HAP和不含AA的HAP相比,在Ser,Asp和Glu存在下制备的HAP均具有较低的结晶度和较高的溶解度。HAP与这些小分子AA结合而产生的特殊性质,有可能模拟早期釉质龋再矿化过程中的无机-有机相互作用,为釉质龋表面和深层的双重修复提供一种简单、经济且有效的方法。
在对单一AA改性HAP研究的基础上,采用化学均相沉淀法,在不同浓度的Ser,Asp和Glu组合下制备AA/HAP复合材料。对AA/HAP复合材料的成分和结构等进行表征,考察其对酸蚀牛牙釉质体外再矿化的影响。对再矿化后的牛牙釉质表面和剖面形貌进行表征,以评价不同复合材料的再矿化效果,并讨论AA/HAP复合材料的再矿化机制。
1 实验材料与方法 1.1 实验材料NaN3,分析纯,福晨(天津)化学试剂有限公司;氨水,分析纯,广东光华科技股份有限公司;其他试剂均为分析纯,国药集团化学试剂有限公司;去离子水,自制。
1.2 氨基酸/羟基磷灰石复合材料的制备采用化学均相沉淀法,选用如表 1所示的3种浓度条件制备AA/HAP复合材料,并将其分别命名为HAP-0,HAP-5和HAP-10。将Ca(NO3)2·4H2O(14.169 g,60 mmol)和不同浓度的Ser,Asp和Glu(由反应溶液的总体积计算的摩尔分数)溶解在1600 mL去离子水中。将混合物加热并在随后的实验过程中保持在(37±1) ℃。将(NH4)2HPO4(4.752 g,35.98 mmol)溶于800 mL去离子水中,并在约120 min内缓慢滴定到混合物中。采用2.5%的氨水调节反应溶液的pH值保持在10±0.5。滴定完成后,继续保温反应24 h。然后将悬浮液常温静置老化24 h,用去离子水洗涤直至pH降至约7.0。最后将沉淀物过滤、冻干后获得AA/HAP复合材料。
| Composite | Ser/ (mmol·L-1) |
Asp/ (mmol·L-1) |
Glu/ (mmol·L-1) |
| HAP-0 | 0 | 0 | 0 |
| HAP-5 | 5 | 5 | 5 |
| HAP-10 | 10 | 10 | 10 |
选用2~3岁龄、完整且无龋损的黄牛磨牙,用刀片除去有机污物并切下根部。然后用DTQ-5低速精密切割机切成3 mm×4 mm×2 mm的釉质块。使用PMMA树脂包埋釉质块,将暴露的釉质唇在水冷金刚砂盘上抛光,直至获得镜面状表面,经体视显微镜检查无明显划痕。将牙釉质样品超声处理1 min,浸入含有乳酸(0.1 mol·L-1)的3%羧甲基纤维素凝胶(pH=4.0)中,脱矿24 h以生成人工龋损。之后用去离子水冲洗样品并超声处理1 min,随后储存在4 ℃的PBS中备用。
1.4 CCK-8法测定细胞毒性采用CCK-8法,测定所制备材料对第3代293T细胞的毒性。制备待测材料浓度为0.05~0.4 g·mL-1的浸提液,并与密度为5.0×104个/孔的293T细胞共培养48 h。随后将10 μL CCK-8溶液加入每个孔中,继续温育4 h,在450 nm波长下测量每个孔的光密度(OD)。相对细胞活性(RCV)按如下公式计算:

|
(1) |
式中:ODt,ODn和OD0分别是含有材料浸提液的孔,不含材料的孔(阴性对照)和没有细胞的孔(空白对照)的平均光密度值。每种材料浓度测量8个孔的OD值,并使用平均值±标准偏差表示。
1.5 体外再矿化实验按Wu等[6]报道的方法制备人工唾液。将牙釉质样品浸入含有60 mL人工唾液和2.16 g再矿化材料(分别为HAP-0,HAP-5和HAP-10)的锥形烧瓶中,使牙釉质抛光面朝上,然后将锥形瓶置于恒温摇床(37 ℃)震荡12 h。之后取出牙釉质样品,用去离子水冲洗20 s,置于37 ℃的新鲜人工唾液中继续浸泡12 h。依此循环共进行21 d,每7 d更换材料悬浮液。空白组:实验流程同实验组,将牙釉质样品分别置于37 ℃的人工唾液中(每7 d更换人工唾液)和新鲜人工唾液中各浸泡12 h,依此循环共进行21 d。
1.6 力学性能分析使用THV-30MD型维氏显微硬度测试仪,测量9.8 N载荷下牙釉质样品的表面显微硬度(SMH),保荷时间为15 s。在不同时间点(脱矿前MH0,脱矿后MH1和再矿化后MH2),测量每个样品表面相距200~300 μm的3个点并记录平均值。将脱矿后样品的SMH从大到小排列,根据SMH值平衡分布原理将样品均匀分成4组(n=6)。表面显微硬度增加率(MHR)按如下公式计算:

|
(2) |
使用SK250HP型超声仪对牙釉质再矿化层的结合力进行检测,功率100 W,超声时间15 s。
1.7 检测与表征采用EQUINOX-55型红外光谱仪在反射模式下进行操作,测定样品的红外光谱,记录波数范围为400~4000 cm-1。采用PANalytical Empyrean型X射线衍射仪分析复合材料的晶体结构,测试条件为CuKα,波长λ=0.15406 nm,步长0.02°,扫描速率4 (°)/min,电压40 kV, 电流40 mA。通过JEM-2100型透射电镜表征复合材料的微观结构特征,加速电压200 kV。采用FEI Nova NanoSEM 450型场发射扫描电镜表征抛光、酸蚀和再矿化后牙釉质样品的表面和剖面形貌。通过电感耦合等离子体发射光谱仪(ICP-OES)测定含有复合材料再矿化溶液的上清液中的Ca2+浓度。
2 结果与分析 2.1 AA/HAP复合材料的表征与分析图 1为HAP-0,HAP-5和HAP-10的红外光谱图(FTIR)。由图 1可以看出,962 cm-1和1094/1040 cm-1分别为PO43-的v1和v3伸缩振动峰,473 cm-1和565/603 cm-1则分别为PO43-的v2和v4弯曲振动峰,3442 cm-1和1633 cm-1分别属于—OH的伸缩振动和吸附水的弯曲振动,证明3种材料均为HAP。HAP-0在1454/874 cm-1和1422 cm-1处的吸收峰分别对应于CO32-的弯曲模式和拉伸模式,说明空气中的CO2溶解在溶液中并参与了化学反应。AA参与了HAP-5和HAP-10的晶体生长过程,因此其1400~1430 cm-1和1450~1485 cm-1处的吸收峰也分别对应于—COO-和—NH3+的对称/不对称峰[16],证实了化学键合AA的存在。并且随着3种AA的浓度从5 mmol·L-1增加到10 mmol·L-1,此处吸收峰强度变得更高。表明随着AA含量增加,HAP晶体中化学键合的AA的相对含量也更高。

|
图 1 HAP-0,HAP-5和HAP-10的FTIR图 Fig. 1 FTIR spectra of HAP-0, HAP-5 and HAP-10 |
图 2为HAP复合材料的X射线衍射图(XRD)。由图 2可知,3种材料的(002),(112),(310),(222),(213),(004)和(304)衍射峰均对应于HAP标准卡索引(JCPDS 09-0432)的特征峰。同时,在HAP的特征衍射峰2θ=31°~33°处明显宽化,表明合成的材料均为纳米HAP。材料的晶粒尺寸D(nm)可通过Debye-Scherer公式计算:
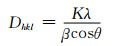
|
(3) |

|
图 2 HAP-0,HAP-5和HAP-10的XRD图 Fig. 2 XRD patterns of HAP-0, HAP-5 and HAP-10 |
式中:K是Scherer常数(0.89);λ是X射线波长(0.15406 nm);θ是衍射角;β是半高全宽。表 2为3种材料沿不同晶向的晶粒尺寸数据。由表 2可知,这3种材料沿c轴方向的晶粒尺寸(即垂直于(002)晶面方向)均为20 nm左右,而沿垂直于(202)晶面方向的尺寸则随着AA用量的增加而不断增大。
| Composite | D/nm | ||
| (002) | (112) | (202) | |
| HAP-0 | 21.05 | 9.13 | 12.39 |
| HAP-5 | 18.40 | 10.14 | 16.47 |
| HAP-10 | 22.33 | 8.55 | 19.39 |
Ser,Asp或Glu单独参与HAP的制备时,所得材料的XRD均显示出合并的特征峰[15],但其衍射峰强度与不含AA的HAP之间没有明显差异。本研究选用Ser,Asp和Glu复合物改变复合材料的衍射峰强度。HAP-5和HAP-10的衍射峰强度明显低于HAP-0,且在(310),(222)和(304)等晶面处明显宽化。表明在复合AA的抑制作用下,HAP在多个晶面方向均发生了尺寸细化。Ser的端羟基可以吸附在HAP晶体表面的缺陷部位,从而使HAP的结晶度降低、溶解度增加[15, 17]。α-羧基和侧末端羧基赋予Asp和Glu在HAP晶体表面上的强亲和力,并显著抑制垂直于(310)晶面的晶体生长,表明酸性AA优先与平行于c轴的晶面作用,使晶体沿[100]方向的生长受到明显抑制[18]。图 3为再矿化溶液上清液中的Ca2+浓度图。由图 3可以看出,含有AA改性HAP的再矿化溶液其Ca2+浓度在7 d的矿化周期内逐渐增加,且随AA用量增加Ca2+浓度相应提高,而含HAP的再矿化液Ca2+浓度呈逐渐降低的趋势。总之,HAP-5和HAP-10晶粒尺寸的细化以及相应的溶解度的增加,可有助于维持再矿化过程中钙磷离子的过饱和状态。

|
图 3 再矿化溶液上清液中Ca2+浓度 Fig. 3 Ca2+ concentration in the supernatant of remineralization solution |
图 4为HAP-0,HAP-5和HAP-10的透射电镜图(TEM)和高分辨透射电镜图(HRTEM)。3种材料均具有类似于牙釉质棱柱的针状形貌。HAP-0的晶粒尺寸(图 4(a-1))为长50~120 nm、宽8~10 nm。而HAP-5(图 4(b-1))和HAP-10(图 4(c-1))的晶粒尺寸均为长70~120 nm、宽3~6 nm。与根据XRD数据计算的晶粒尺寸相比,较长的晶粒尺寸可能是因为TEM所示晶体是由微小的晶界叠加而成的。在复合AA存在下,HAP纳米颗粒的宽度尺寸减小,这一结果进一步证实了图 3所示的Asp和Glu对(310)晶面的抑制作用。此外,HAP-10晶粒的尺寸分布比HAP-5更均匀,这是因为随着AA浓度的增加,其调节HAP晶粒沿[001]晶向生长的效率得到提高。

|
图 4 复合材料的TEM(1)与HRTEM(2)图 (a)HAP-0;(b)HAP-5;(c)HAP-10 Fig. 4 TEM(1) and HRTEM(2) images of the composites (a)HAP-0;(b)HAP-5;(c)HAP-10 |
为了进一步详细研究晶体结构,采用HRTEM表征HAP晶体的晶格条纹。由图 4(a-2)可以看出,重复的晶面间距d=0.339 nm对应于HAP-0的(002)面,表明[001]是优选的晶体生长方向[19]。除(002)面之外,HAP-5的HRTEM图中的晶面间距d=0.284 nm和d=0.253 nm(图 4(b-2)),分别对应于(211)和(301)面。图 4(c-2)中所示的晶面间距d=0.363 nm和d=0.264 nm,分别对应HAP-10中的(201)和(202)面。图 4(b-2),(c-2)背景中直径约为1.5~2.5 nm的黑点可能与HAP-5和HAP-10晶体中捕获的AA炭化有关。因此,TEM和HRTEM图显示不同浓度的AA使HAP晶体的结构完整性降低并产生了细化的晶粒尺寸。
图 5为3种材料的细胞毒性实验结果。参照GB/T16886.5—2017,实验浓度范围内HAP-0的细胞毒性为0级或1级。而HAP-5和HAP-10这两种复合材料的细胞毒性在较宽的浓度范围内均为0级,且随着AA用量的增加,RCV值进一步提高。因此,AA的引入有利于提高纳米HAP材料的生物相容性。

|
图 5 复合材料对细胞相对活性的影响 Fig. 5 Effect of the composites on relative cells viability |
图 6为抛光和酸蚀牙釉质样品的场发射扫描电镜图(FESEM)。由图 6(a-1)可以看出,抛光牙釉质表面致密,其剖面也较完整(图 6(a-2))。酸蚀牙釉质表面呈鳞片状,在相邻釉柱之间形成了明显间隙(图 6(b-1)),且在高放大倍数下可清晰地观察到牙釉质棱晶。牙釉柱的侧面杆鞘处具有疏水性,可以保护牙釉柱免受酸蚀[10]。但釉柱间矿物含有少量有机残余物且结晶度较低,较牙釉柱更易受到酸蚀,最终发展为较大间隙。由图 6(b-2)可以看出,酸蚀牙釉质的剖面具有深达30~50 μm的表面下脱矿形貌。与表层牙釉质相比,深层牙釉质中的晶体尺寸变小、结晶度降低,同时有机残留物和碳酸盐的含量逐渐增加。所有这些特征都使深层牙釉质产生了更严重的脱矿现象。

|
图 6 牙釉质表面(1)和剖面(2)FESEM图 (a)抛光牙釉质;(b)酸蚀牙釉质 Fig. 6 Surface(1) and cross-section(2) FESEM images of the enamel (a)polished enamel; (b)acid-etched enamel |
图 7为酸蚀牙釉质再矿化后的FESEM图。由图 7可以看出,所有实验组的牙釉质表面缺陷均得到了一定程度的修复。空白组(图 7(a-1))表面平整而致密,这归因于牙釉质的自我修复能力。图 7(b-1)所示形貌表明HAP-0主要利用纳米颗粒的吸附特性,从而在釉质表面的空腔中形成不紧密的矿化层。由图 7(c-1)和图 7(d-1)(可见,HAP-5和HAP-10处理组均可在牙釉质表面上生成一层致密的矿物层。在Ser,Asp和Glu存在下制备的HAP复合材料具有更加无序的晶体结构和更高的溶解度[15],因此可提供更多的Ca2+和PO43-用于牙釉质样品的表面再矿化。同时,吸附在牙釉质表面的AA分子可以吸附Ca2+和PO43-并维持局部过饱和[20],最终促进新形成的HAP晶体的生长。

|
图 7 再矿化后牙釉质表面(1)和剖面(2)FESEM图 (a)空白组;(b)HAP-0;(c)HAP-5;(d)HAP-10 Fig. 7 Surface(1) and cross-section(2) FESEM images of the enamel after remineralization (a)blank group; (b)HAP-0;(c)HAP-5;(d)HAP-10 |
空白组的剖面形貌显示未修复的釉柱间缺陷仍然存在(图 7(a-2))。HAP-0的剖面图(图 7(b-2))则表现出严重的釉柱间缺陷,牙釉质深层的未修复区域比表层更明显。如XRD图中所分析(图 2),HAP-0具有较高的结晶度,虽然其在牙釉质表层修复中有一定效果,但在深层修复中无效。同时,HAP-0晶体可以吸附Ca2+和PO43-并在37 ℃的人工唾液中继续生长,因此导致其对牙釉质深层的再矿化效果比空白组差。HAP-5的深层再矿化作用(图 7(c-2))优于HAP-0,但与空白组相比差异不大,这可能与再矿化过程中释放的Ser,Asp和Glu浓度较低有关。HAP-10的剖面图(图 7(d-2))显示已生成一层厚度约为22 μm的致密且均匀的再矿化层。尽管在AA/HAP复合材料的制备过程中,反应溶液中存在的Ser,Asp和Glu会抑制HAP的晶体生长,但再矿化过程中从HAP释放的AA则可以促进牙釉质龋损的修复。AA分子尺寸较小,比较容易渗透到深层脱矿区域中。当AA分子吸附在有机基质的残基上后,可以作为形核位点从而促进致密矿化层的形成。
图 8为牙釉质样品表面的力学性能结果。配对样品T检验结果显示,空白组再矿化前后SMH无显著差异(p>0.05),所有材料处理组再矿化前后的SMH之间均存在显著差异(p < 0.05),而HAP-10处理的样品具有最高的SMH值。酸蚀处理后,牙釉质样品SMH的均值从(274.77±15.53)HV(抛光牙釉质)降低至约27HV。研究发现,牙釉质的韧性大约是地质HAP的3倍[21]。牙釉质具有复杂的分级结构,并通过牙釉质棱柱、牙釉柱和有机残余物的精确排列得到加强[2],一旦被侵蚀到内部区域,这种精细的结构就会被破坏,牙釉质的韧性和硬度就无法完全恢复。力学性能测试结果显示,HAP-0,HAP-5和HAP-10组的MHR分别为35.11%,29.63%和54.45%。因此,尽管HAP-10组获得了最佳的SMH恢复效果,但3种材料再矿化后的SMH互相之间无显著差异(p>0.05)。另外,经超声处理后,空白组和3个实验组表面再矿化层均无明显脱落现象,说明再矿化层结合力良好。

|
图 8 再矿化前后SMH值对比图 Fig. 8 Comparison of the SMH values before and after remineralization |
(1) 通过化学均相沉淀法制备了Ser,Asp和Glu改性的AA/HAP复合材料。AA使AA/HAP的形核和晶面生长受到干扰,显著抑制垂直于(310)面的晶体生长,从而使晶体颗粒宽度变小,并导致HAP的溶解度增加和晶体结构有序性降低。HRTEM晶格分析显示AA/HAP颗粒呈现出(211)或(201)等许多短程的晶面,证明在AA的修饰下HAP的晶粒尺寸产生了细化。
(2) 细胞毒性结果表明,AA/HAP复合材料的相对细胞活性明显高于HAP,说明AA的引入有利于提高AA/HAP复合材料的生物相容性。
(3) 体外再矿化结果显示,AA/HAP复合材料可在酸蚀牛牙釉质表面生成一层致密的再矿化层,而HAP仅能部分吸附和填充在牙釉质表面的空腔中。在3种AA浓度为10 mmol·L-1条件下制备的复合材料,可在深层龋损区域生成厚度约为22 μm的致密再矿化层,并获得最佳的表面显微硬度恢复效果。
| [1] |
李小科, 王进防, 常江. 牙釉质再矿化综述[J]. 口腔护理用品工业, 2017, 27(2): 18-26. LI X K, WANG J F, CHANG J, et al. The remineralisation of enamel: a review of the literature[J]. Oral Care Industry, 2017, 27(2): 18-26. |
| [2] |
HE L H, SWAIN M V. Understanding the mechanical behaviour of human enamel from its structural and compositional characteristics[J]. Journal of the Mechanical Behavior of Biomedical Materials, 2008, 1(1): 18-29. DOI:10.1016/j.jmbbm.2007.05.001 |
| [3] |
FAN Y, NELSON J R, ALVAREZ J R, et al. Amelogenin-assisted ex vivo remineralization of human enamel: effects of supersaturation degree and fluoride concentration[J]. Acta Biomaterialia, 2011, 7(5): 2293-2302. DOI:10.1016/j.actbio.2011.01.028 |
| [4] |
LI Q L, NING T Y, CAO Y, et al. A novel self-assembled oligopeptide amphiphile for biomimetic mineralization of enamel[J]. BMC Biotechnology, 2014, 14(1): 1-11. DOI:10.1186/1472-6750-14-1 |
| [5] |
LE N E, KWAK S Y, WIEDEMANNBIDLACK F B, et al. Leucine-rich amelogenin peptides regulate mineralization in vitro[J]. Journal of Dental Research, 2011, 90(9): 1091-1097. DOI:10.1177/0022034511411301 |
| [6] |
WU D, YANG J, LI J, et al. Hydroxyapatite-anchored dendrimer for in situ remineralization of human tooth enamel[J]. Biomaterials, 2013, 34(21): 5036-5047. DOI:10.1016/j.biomaterials.2013.03.053 |
| [7] |
毛文文, 茹江英. 羟基磷灰石类陶瓷在骨组织工程中的研究与更广泛应用[J]. 中国组织工程研究, 2018, 22(30): 4855-4863. MAO W W, RU J Y. Hydroxyapatite ceramics in bone tissue engineering: research and extensive applications[J]. Chinese Journal of Tissue Engineering Research, 2018, 22(30): 4855-4863. |
| [8] |
LELLI M, PUTIGNANO A, MARCHETTI M, et al. Remineralization and repair of enamel surface by biomimetic Zn-carbonate hydroxyapatite containing toothpaste: a comparative in vivo study[J]. Frontiers in Physiology, 2014, 5: 1-7. |
| [9] |
VENEGAS S C, PALACIOS J M, APELLA M C, et al. Calcium modulates interactions between bacteria and hydroxyapatite[J]. Journal of Dental Research, 2006, 85(12): 1124-1128. DOI:10.1177/154405910608501211 |
| [10] |
DUVERGER O, BENIASH E, MORASSO M I. Keratins as components of the enamel organic matrix[J]. Matrix Biology, 2016, 52/54: 260-265. DOI:10.1016/j.matbio.2015.12.007 |
| [11] |
WIEDEMANN-BIDLACK F B, KWAK S Y, BENIASH E, et al. Effects of phosphorylation on the self-assembly of native full-length porcine amelogenin and its regulation of calcium phosphate formation in vitro[J]. Journal of Structural Biology, 2011, 173(2): 250-260. DOI:10.1016/j.jsb.2010.11.006 |
| [12] |
LI L, MAO C, WANG J, et al. Bio-inspired enamel repair via glu-directed assembly of apatite nanoparticles: an approach to biomaterials with optimal characteristics[J]. Advanced Materials, 2011, 23(40): 4695-4701. DOI:10.1002/adma.201102773 |
| [13] |
WU X, ZHAO X, LI Y, et al. In situ synthesis carbonated hydroxyapatite layers on enamel slices with acidic amino acids by a novel two-step method[J]. Materials Science and Engineering: C, 2015, 54: 150-157. DOI:10.1016/j.msec.2015.05.006 |
| [14] |
FAN Z J, WANG J Q, WANG Z F, et al. One-pot synthesis of graphene/hydroxyapatite nanorod composite for tissue engineering[J]. Carbon, 2014, 66(1): 407-416. |
| [15] |
MATSUMOTO T, OKAZAKI M, INOUE M, et al. Crystallinity and solubility characteristics of hydroxyapatite adsorbed amino acid[J]. Biomaterials, 2002, 23(10): 2241-2247. DOI:10.1016/S0142-9612(01)00358-1 |
| [16] |
GONZALEZ-MCQUIRE R, CHANE-CHING J Y, VIGNAUD E, et al. Synthesis and characterization of amino acid-functionalized hydroxyapatite nanorods[J]. Journal of Materials Chemistry, 2004, 14(14): 2277-2281. DOI:10.1039/b400317a |
| [17] |
JACK K S, VIZCARRA T G, TRAU M. Characterization and surface properties of amino-acid-modified carbonate-containing hydroxyapatite particles[J]. Langmuir, 2007, 23(24): 12233-12242. DOI:10.1021/la701848c |
| [18] |
BOANINI E, TORRICELLI P, GAZZANO M, et al. Nanocomposites of hydroxyapatite with aspartic acid and glutamic acid and their interaction with osteoblast-like cells[J]. Biomaterials, 2006, 27(25): 4428-4433. DOI:10.1016/j.biomaterials.2006.04.019 |
| [19] |
PARK S Y, KIM K I, PARK S P, et al. Aspartic acid-assisted synthesis of multifunctional strontium-substituted hydroxyapatite microspheres[J]. Crystal Growth & Design, 2016, 16(8): 4318-4326. |
| [20] |
MATSUMOTO T, OKAZAKI M, INOUE M, et al. Role of acidic amino acid for regulating hydroxyapatite crystal growth[J]. Dental Materials Journal, 2006, 25(2): 360-364. DOI:10.4012/dmj.25.360 |
| [21] |
WHITE S N, LUO W, PAINE M L, et al. Biological organization of hydroxyapatite crystallites into a fibrous continuum toughens and controls anisotropy in human enamel[J]. Journal of Dental Research, 2001, 80(1): 321-326. DOI:10.1177/00220345010800010501 |