 2020, Vol. 48
2020, Vol. 48文章信息
- 焦华, 赵康, 石蕊, 马利宁, 卞铁荣, 汤玉斐
- JIAO Hua, ZHAO Kang, SHI Rui, MA Li-ning, BIAN Tie-rong, TANG Yu-fei
- 羟基磷灰石纳米棒的水热制备及其晶体生长机理研究
- Hydrothermal synthesis and crystal growth mechanism of hydroxyapatite nanorods
- 材料工程, 2020, 48(1): 136-143
- Journal of Materials Engineering, 2020, 48(1): 136-143.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2018.001206
-
文章历史
- 收稿日期: 2018-10-15
- 修订日期: 2019-09-26




在生物医学材料领域,纳米材料及其结构的研究引起了各国学者的广泛关注。生物体内存在着大量的具有特殊功能的纳米结构,如在骨骼、牙齿、肌腱等器官,均不同程度存在规则分级的纳米组装结构。生物材料中的磷灰石,尤其是纳米羟基磷灰石和天然骨中的羟基磷灰石(Ca10(PO4)6(OH)2,Hydroxyapatite,HA)在化学组成、结构等方面极其相似。因此,羟基磷灰石纳米材料以其良好的生物相容性和骨传导能力在硬组织的替代和填充方面得到了广泛的研究和应用[1-2]。HA是人体骨的主要成分,也有人把HA称为人工骨。它的特点是:生物活性及其相容性优越、不存在免疫排斥反应、无毒,没有致癌致死性,并且在体内降解后能够与骨直接结合,具有较好的骨传导性,因此被认为是一种最具潜力的人体硬组织替代材料[3-4]。另一方面HA材料具有生物相容性和生物活性,符合骨组织工程对材料的生物学要求[5]。目前的研究结果表明,纳米羟基磷灰石能够促使新骨大量形成,生物降解性以及骨引导性均良好[6]。吸附是HA载药的主要方式,具有一维微纳米结构的HA材料,相比颗粒会暴露出更多的活性位点,更能满足各种生物活性物质的高载药量需求,而成为骨组织工程的一个重要研究方向。微纳米尺度的表面在一维HA材料中的联合作用不仅有利于细胞增殖和成骨分化,而且有利于血管生成因子在干细胞分化中的表达[7]。因此,制备得到单晶一维微纳米结构的HA在生物医药领域的研究方面具有重要的意义。
由于羟基磷灰石在高温时容易脱羟基,所以HA多采用液相合成法。通常用于制备形貌可控HA的方法有:水热法[8-11]、模板法[12]、微乳液法[13]、微波法[14]和超声法[15]。固相反应法往往给出符合化学计量、结晶完整的产品,不足之处是要求较高的温度和热处理时间;水热合成法获得的HA材料一般结晶程度较高,钙磷比接近化学计量比。本工作采用水热法,以无水CaCl2和(NH4)2HPO4为原料,尿素为均相沉淀剂,十六烷基三甲基溴化铵(CTAB)为模板剂,制备HA纳米棒,主要研究了不同水热温度及时间变化对HA形貌、尺寸和相组成等的影响,得出在120 ℃反应12 h为最佳工艺条件。从晶体结构的角度出发,详细探讨了CTAB在HA纳米棒形成过程中所起的作用。
1 实验材料及方法 1.1 原料与仪器无水氯化钙(CaCl2,AR,天津化学试剂厂);磷酸氢二铵((NH4)2HPO4,AR,成都化学试剂厂);氢氧化钠(NaOH,AR,西安化学试剂厂);尿素(H2NCONH2,AR,天津化学试剂厂);十六烷基三甲基溴化铵((C16H33 (CH3)3NCr),CTAB,AR,西安化学试剂厂);去离子水。
1.2 实验过程称取0.78 g无水CaCl2,0.55 g (NH4)2HPO4,分别置于盛有35 mL去离子水的烧杯中,搅拌溶解,得到按照Ca/P原子比为1.67配置的溶液。然后将二者混合并搅拌,出现白色乳浊液。再称取0.2 g CTAB及1.5 g尿素依次加入混合溶液中,持续搅拌15 min后将最终混合后的悬浮液倒入100 mL高压反应釜中,分别于90~180 ℃下水热处理4~16 h,冷却至室温,离心分离,用去离子水及无水乙醇将白色沉淀洗涤3次后于60 ℃条件下干燥5 h,得到产物。
1.3 样品表征采用XRD-7000 X射线衍射仪分析产物的物相组成,测试条件为CuKα辐射,最大管电压60 kV,最大管流80 mA,扫描速率8 (°) / min。采用JEM-6700F扫描电镜观察产物的微观形貌。采用JEM-3010高分辨透射电镜(HRTEM)表征产物的微观结构,加速电压为200 kV。
2 结果与讨论 2.1 不同水热温度下产物的XRD分析图 1为反应物在90,120,140 ℃和180 ℃下水热反应12 h所得产物的XRD图。由图 1可知,在水热温度较低为90 ℃时,产物中存在少量的杂质峰与正交晶系CaCO3 (PDF No.41-1475)的(111)晶面和六方晶系(Ca, Mn)CO3 (PDF No.02-0714)的(104)晶面相对应。在较低温度90 ℃反应时,由于所用原料中的尿素在水中会存在CO32-,推测其主要与Ca2+发生了反应生成了杂质相。而随着反应温度的升高,尿素产生的主要为CO2和OH-,随着体系中OH-越来越多,Ca2+与PO43-和OH-的过饱和度越大,HA成核越易,因此在其他温度下120~180 ℃的衍射峰与六方晶系HA的标准卡片(PDF No.09-0432)中的衍射峰一一对应,没有杂质相的存在,均可得到单一的HA产物。

|
图 1 不同反应温度下水热反应12 h所得产物的XRD图 Fig. 1 XRD patterns of samples after hydrothermal at different reaction temperatures for 12 h |
表 1为羟基磷灰石样品在不同温度90~180 ℃反应12 h的晶面指数和强度因子。由表 1可以看出,实际样品测试的XRD数据得出该样品的(002)晶面的强度I(002)与(211)晶面的强度I(211),通过计算两个晶面的相对比值I(002)/I(211),可以与标准卡片(PDF No.09-0432)的数据I(002)=40,I(211)=100计算的I(002)/I(211)=0.400进行对比,得出当水热温度在120~180 ℃范围内时,c轴上晶粒尺寸均大于a轴方向上的晶粒尺寸。表明HA纳米棒是平行于c轴方向生长。在120 ℃反应12 h所得样品的(002)晶面方向上的衍射峰相对强度最强。说明在此条件下,所得样品的长径比最大。
| Temperature/℃ | I(002) | I(211) | I(002)/I(211) |
| 90 | 32 | 88 | 0.363 |
| 120 | 152 | 180 | 0.844 |
| 140 | 56 | 108 | 0.519 |
| 180 | 58 | 94 | 0.617 |
图 2为反应物在90,120,140 ℃和180 ℃下水热反应12 h所得产物的SEM照片。由图 2可以看出,在不同水热温度下,均生成了棒状结构的HA。在90 ℃的水热温度下,产物结晶度差,合成的棒状结构也不完整,尺寸较大且表面粗糙(图 2(a))。从图 2(b)可以看出,升高温度到120 ℃时,产物的形貌为棒状结构,尺寸较为均匀,直径约为20~30 nm,长度在0.5~0.8 μm。当温度升高到140 ℃时(图 2(c)),生成的HA纳米棒直径范围是20~100 nm,变化幅度比较大。当温度继续升高到180 ℃时,生成的HA纳米棒长度明显变短,平均为300 nm(图 2(d)),这说明较高的反应温度时,得到长度较好,直径分布均匀的HA并不有利。因此得出120 ℃为合适的反应温度。

|
图 2 不同水热温度下反应12h所得产物的SEM图 (a)90 ℃; (b)120 ℃; (c)140 ℃; (d)180 ℃ Fig. 2 SEM images of samples after hydrothermal at different reaction temperatures for 12 h (a)90 ℃; (b)120 ℃; (c)140 ℃; (d)180 ℃ |
实验对120 ℃,不同水热时间4,8,12 h及16 h的产物进行了XRD分析,如图 3所示。与标准卡片(PDF No.09-0432)进行对比分析,结果表明,当反应时间过短时,水热时间4 h,如图 3中曲线所示,产物不是纯的HA相,有部分杂质相正交晶系CaCO3(PDF No.41-1475)和六方晶系的(Ca, Mn)CO3 (PDF No.02-0714)。之后随着反应时间的延长超过8 h,杂质相逐渐减少。当时间延长到12 h时,即可得到纯的HA物质,且有良好的结晶度。继续延长反应时间到16 h,产物的结晶化程度有略微增加但变化并不明显。

|
图 3 120 ℃水热下不同反应时间所得产物的XRD图 Fig. 3 XRD patterns of samples after hydrothermal at 120 ℃ for different reaction time |
表 2为羟基磷灰石样品在120 ℃下反应不同时间的晶面指数和晶面强度。由表 2可见,实际样品测试的XRD数据得出该样品的(002)晶面的强度I(002)与(211)晶面的强度I(211),通过计算两个晶面的相对比值I(002)/I(211),与标准卡片的数据进行对比,得出当水热温度在8~16 h范围内,I(002)/I(211)的比值均大于标准卡片上的比值,表明HA纳米棒是沿着(002)晶面取向生长,平行于c轴方向。在120 ℃反应12 h所得样品的(002)晶面方向上的衍射峰相对强度最强,说明在该条件下纳米棒的长径比最大。
| Time/h | I(002) | I(211) | I(002)/I(211) |
| 4 | 72 | 206 | 0.349 |
| 8 | 76 | 96 | 0.792 |
| 12 | 152 | 180 | 0.844 |
| 16 | 124 | 200 | 0.620 |
选择120 ℃研究不同水热时间下的产物。图 4为反应物在120 ℃不同水热时间4,8,12 h及16 h下所得产物的SEM照片。由图 4可以看出,水热时间的变化不会导致棒状结构的明显改变,产物的形貌仍为HA纳米棒。但水热时间对HA纳米棒的尺寸形成了一定的影响。当反应时间从4 h逐渐增加到16 h时,纳米棒的直径有缓慢增大的趋势,这是因为时间的延长有利于晶核的形成和长大,使晶粒发育完整,从而使棒的直径呈缓慢增加的趋势。但反应时间过短或过长都会导致棒状尺寸不均匀。当反应时间过短时,如图 4(a),(b)所示,棒状HA没有充分生长,组织内部存在择优生长现象,就会导致棒状结构的直径与长度等尺寸范围分布较广。而当反应时间过长时,如图 4(d)所示,HA棒状结构直径变大为50~80 nm,还有部分纳米棒断裂破碎。结合XRD的数据结果,综合分析得出,本实验制备HA纳米棒最佳水热条件为:120 ℃,12 h。

|
图 4 120 ℃水热下不同反应时间所得产物的SEM照片 (a)4 h; (b)8 h; (c)12 h; (d)16 h Fig. 4 SEM images of samples after hydrothermal at 120 ℃ for different reaction time (a)4 h; (b)8 h; (c)12 h; (d)16 h |
对反应物在120 ℃水热反应12 h所得产物进行进一步的结构分析,如图 5所示。HA纳米棒代表性的TEM图像和SAED图,沿[010]晶带轴进行收集。由图 5(a)可以看出,HA纳米棒状结构尺寸分布均匀,直径约为20 nm,长度范围为0.5~0.8 μm与SEM的结果基本一致。HA纳米棒的结晶生长方向平行于c轴[001]方向,图 5(b)中的插图为HA晶体的模型,由图可以看出,六方晶体HA纳米棒的结晶生长方向平行于c轴[001]方向。对纳米棒进行选区电子衍射SAED分析,结果见图 5(c),图中衍射斑点清晰明亮,呈现阵列整齐排列,表现出典型的单晶结构特征。图 5(d)为图 5(b)矩形框中选区的HRTEM照片,晶面纹路清晰,经过测量计算,晶面间距约为0.344 nm,对应于密排六方相HA的(002)晶面间距值。由以上结果得出,120 ℃水热反应12 h所得产物为单晶六方相结构的HA纳米棒。

|
图 5 120 ℃, 12 h水热条件下所得产物的TEM(a),单根HA纳米棒的TEM和晶体模型(b),SAED图(c),HRTEM图(d) Fig. 5 TEM(a), single HA nanorod and crystal model(b), SAED(c) and HRTEM(d) images of samples after hydrothermal treatments at 120 ℃ for 12 h |
为了进一步确定产物的组分,对120 ℃水热反应12 h所得产物进行了红外光谱分析见图 6。由图 6中曲线可见,红外光谱的吸收带在3437 cm-1和1646 cm-1处归因于吸附的H2O的弯曲模式[16]。吸收带在607 cm-1和565 cm-1归因于PO43-的弯曲振动模式[14],1038 cm-1和960 cm-1与PO43-的拉伸振动模式有关[17]。此外,CO32-的吸收带在1465,1416 cm-1和872 cm-1可以观察到[18],这与物质中加入尿素会释放出CO2部分溶解于溶液中形成CO32-有关。OH-的伸缩振动模式在3570 cm-1可以观察到。上述结果证明,HA的官能团均在红外光谱上有显示,由此得出,在该条件下成功地合成了HA。

|
图 6 120 ℃水热12 h所得产物的IR图 Fig. 6 IR pattern of samples after hydrothermal at 120 ℃ for 12 h |
在水热反应中,随着反应温度的升高,尿素作为均相沉淀剂,发生分解,在溶液中提供了OH-,为生成羟基磷灰石提供了碱性环境,反应方程如下:
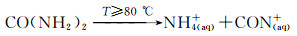
|
(1) |
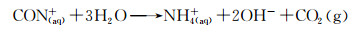
|
(2) |
随着反应的进行,OH-越来越多,溶液的pH值增大,HA的过饱和度越大,越易成核并长大。CaCl2和(NH4)2HPO4在这样的碱性环境中发生化学反应最终生成羟基磷灰石。
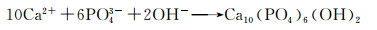
|
(3) |
在HA的生长体系中,由于a轴与c轴方向上的主要生长基元不同,导致生长速率不同。在c轴方向上的生长基元主要为Ca2+,而a轴的主要生长基元为OH-。HA晶体属于六方晶系,是一种L6PC对称的P63/m空间群。晶胞参数为:a=b=0.9423 nm,c=0.6875 nm。a,b轴互成120°夹角,a,b轴与c轴垂直。一个晶胞中含有10个Ca2+,6个PO43+,2个OH-,其晶体结构的模型见图 7(a)。对HA晶面的研究[19]得出,(001)表面是带正电荷,因为在表面主要存在着Ca2+的位置。这种带正电荷的生物陶瓷表面会吸引带负电荷的末端离子、生物活性分子或蛋白质的侧链。而HA的(100)晶面,根据Zhao等[20]的研究,利用分子动力学(MD)模拟来确定纳米晶的HA与水的界面能,HA的(100)和(001)面与水相互作用,(100)面吸水的密度大于(001)面。因此(100)晶面是最有利的亲水面,整个(100)面都是亲水的,水可以相对自由地扩散到表面。当一个分子带电的侧链接近这样一个表面时,界面水的自由能屏障就会降低。这使得侧链能够穿透界面层,直接与表面离子相互作用。其他的晶面如(110)没有亲水性。这些结果为将带电分子吸附到HA表面提供了一个热力学基础。相比之下,非有机生长的磷灰石晶体得到的针状或棒状[12]的形态,主要暴露出来的表面是(100)面,这与图 5透射电镜的分析结果一致。

|
图 7 HA立体结构(a),CTAB结构图(b),HA晶体局部与CTA+结合示意图(c) Fig. 7 HA stereoscopic structure(a), CTAB structure(b) and schematic image of part of HA crystal combined with CTA+(c) |
在水热过程中,CTAB为一种阳离子表面活性剂,CTAB的分子结构如图 7(b)所示,该分子在水溶液中呈长棒状胶束结构,分子是由两个部分组成:一端是带正电的亲水头部与水分子具有较强的亲和力,另一端是疏水尾部与水分子亲和力较弱。CTAB分子在水溶液体系分离成CTA+和Br-,胶束表面带正电的CTA+[21]。在反应过程中,带正电的CTA+可以选择性地吸附在HA(图 7 (a))亲水的(100)晶面上,而此时形成的HA前驱体会继续发生化学反应,使得HA按一定方向依附在胶束表面生成,如图 7(c)所示。因此有效地钝化了HA的亲水的(100)晶面,使平行于c轴[001]方向的晶面增长速率比其他晶面快很多倍。因此,单向增长导致了高度各向异性的纳米棒结构。Lu等[22]曾经报道过当CTAB的胶束浓度大于0.003 mol·L-1时,胶束转变为棒状胶束,本实验中的浓度为0.015 mol·L-1,胶束为棒状结构,因此得到了棒状的羟基磷灰石。
为了进一步证明CTAB对结构的影响,体系中所有反应物的加入顺序和反应浓度等条件不变,在120 ℃水热反应12 h,不加CTAB进行了进一步的SEM和XRD分析,如图 8所示。从图 8(a)可以看出,产物的形貌为不规则颗粒,XRD衍射峰和标准卡片(PDF No.09-0432)的衍射峰一一对应,说明不加CTAB也可以得到六方晶系的HA,但是无法得到棒状结构。因此,SEM照片进一步证明了CTAB的存在对合成棒状结构的HA具有非常关键的作用。

|
图 8 不加CTAB所得产物的SEM图(a)和XRD图(b) Fig. 8 SEM image(a) and XRD pattern(b) of the products without CTAB |
(1) 以无水CaCl2和(NH4)2HPO4为原料,通过水热法制备了HA纳米棒,研究了不同水热温度、水热时间对纳米HA形貌和尺寸的影响。
(2) 制备HA纳米棒的最佳水热条件为:120 ℃,12 h。在此条件下可以制备出单晶的密排六方结构HA纳米棒,直径约为15~30 nm,长度范围为0.5~1.0 μm。
(3) 从晶体结构的角度详细研究了CTAB在合成纳米棒结构中所起的作用,并通过实验进行了验证,所制备的HA纳米棒材料在骨自愈、骨重建和骨再生方面具有广阔的应用前景。
| [1] |
AKRAM M, AHMED R, SHAKIR I, et al. Extracting hydroxyapatite and its precursors from natural resources[J]. Journal of Materials Science, 2014, 49(4): 1461-1475. DOI:10.1007/s10853-013-7864-x |
| [2] |
张平生, 辛勇, 曹传亮, 等. 壳聚糖/羟基磷灰石表面修饰聚己内酯多孔骨支架的制备及性能[J]. 材料工程, 2019, 47(7): 64-70. ZHANG P S, XIN Y, CAO C L, et al. Preparation and properties of polycaprolactone porous bone scaffold modified with chitosan/hydroxyapatite on the surface[J]. Journal of Materials Engineering, 2019, 47(7): 64-70. |
| [3] |
CRANE G M, ISHAUG S L, MIKOS A G. Bone tissue engineering[J]. Nature Medicine, 1995, 1(12): 1322-1326. DOI:10.1038/nm1295-1322 |
| [4] |
JOHNSON A J W, HERSCHLER B A. A review of the mechanical behavior of CaP and CaP/polymer composites for applications in bone replacement and repair[J]. Acta Biomaterialia, 2011, 7(1): 16-30. DOI:10.1016/j.actbio.2010.07.012 |
| [5] |
JR D L W, EINHORN T A, KOVAL K, et al. Bone grafts and bone graft substitutes in orthopaedic trauma surgery[J]. Journal of Bone and Joint Surgery-American Volume, 2007, 89(3): 649-658. DOI:10.2106/00004623-200703000-00026 |
| [6] |
张欣, 孙红. 纳米羟基磷灰石及其复合物修复骨缺损的研究与应用[J]. 中国组织工程研究, 2012, 16(34): 6403-6406. ZHANG X, SUN H. Nano-hydroxyapatite and its compound in repairing bone defects[J]. Chinese Journal of Tissue Engineering Research, 2012, 16(34): 6403-6406. |
| [7] |
STOJANOVIC' Z S, IGNJATOVIC' N, WU V, et al. Hydrothermally processed 1D hydroxyapatite: mechanism of formation and biocompatibility studies[J]. Materials Science & Engineering:C, 2016, 68(1): 746-757. |
| [8] |
ITO H, OAKI Y, IMAI H. Selective synthesis of various nanoscale morphologies of hydroxyapatite various an intermediate phase[J]. Crystal Growth & Design, 2008, 8(3): 1055-1059. |
| [9] |
NEIRA I S, KOLEN'KO Y V, LEBEDEV O I, et al. An effective morphology control of hydroxyapatite crystals via hydrothermal synthesis[J]. Crystal Growth & Design, 2009, 9(1): 466-474. |
| [10] |
ZHANG C M, YANG J, QUAN Z W, et al. Hydroxyapatite nano and microcrystals with multiform morphologies:controllable synthesis and luminescence properties[J]. Crystal Growth & Design, 2009, 9(6): 2725-2733. |
| [11] |
GUO Y P, YAO Y B, NING C Q, et al. Fabrication of mesoporous carbonated hydroxyapatite microspheres by hydrothermal method[J]. Materials Letters, 2011, 65(14): 2205-2208. DOI:10.1016/j.matlet.2011.04.057 |
| [12] |
ZHANG L, ZHU S, HAN Y, et al. Formation and bioactivity of HA nanorods on micro-arc oxidized zirconium[J]. Materials Science & Engineering:C, 2014, 43(8): 86-91. |
| [13] |
KUMAR G S, THAMIZHAVEL A, GIRIJA E K. Microwave conversion of eggshells into flower-like hydroxyapatite nanostructure for biomedical applications[J]. Materials Letters, 2012, 76(6): 198-200. |
| [14] |
SHAVANDI A, BEKHITA E D, AZAM A, et al. Synthesis of nano-hydroxyapatite (nHA) from waste mussel shells using a rapid microwave method[J]. Materials Chemistry and Physics, 2015, 149/150(1): 607-616. |
| [15] |
POINERN G E, BRUNDAVANAM R K, MONDINOS N, et al. Synthesis and characterization of nanohydroxyapatite using an ultrasound assisted method[J]. Ultrasonics Sonochemistry, 2009, 16(4): 469-474. DOI:10.1016/j.ultsonch.2009.01.007 |
| [16] |
KOUTSOPOULOS S. Synthesis and characterization of hydroxyapatite crystals: a review study on the analytical methods[J]. Journal of Biomedical Materials Research, 2002, 62(4): 600-612. DOI:10.1002/jbm.10280 |
| [17] |
KANCHANA P, SEKAR C. Development of electrochemical folic acid sensor based on hydroxyapatite nanoparticles[J]. Spectrochimica Acta Part:A, 2015, 137(2): 58-65. |
| [18] |
WEI D Q, ZHOU Y, YANG C H. Structure, cell response and biomimetic apatite induction of gradient TiO2-based/nano-scale hydrophilic amorphous titanium oxide containing Ca composite coatings before and after crystallization[J]. Colloids and Surfaces:B, 2009, 74(1): 230-237. DOI:10.1016/j.colsurfb.2009.07.025 |
| [19] |
RULIS P, YAO H Z, OUYANG L Z, et al. Electronic structure, bonding, charge distribution, and X-ray absorption spectra of the (001) surfaces of fluorapatite and hydroxyapatite from first principles[J]. Physical Review:B, 2007, 76(24): 5410-5414. |
| [20] |
ZHAO W L, XU Z J, YANG Y, et al. Surface energetics of the hydroxyapatite nanocrystal-water interface: a molecular dynamics study[J]. Langmuir, 2014, 30(44): 13283-13292. DOI:10.1021/la503158p |
| [21] |
LI Y, TJANDRA W, TAM K C. Synthesis and characterization of nanoporous hydroxyapatite using cationic surfactants as templates[J]. Materials Research Bulletin, 2008, 43(8/9): 2318-2326. |
| [22] |
卢惠娟, 陈冲, 郭宏涛, 等. 无探针紫外光谱法测定CTAB的第二临界胶束浓度[J]. 化学学报, 2006, 64(24): 2437-2441. LU H J, CHEN C, GUO H T, et al. Determination of the second critical micelle concentration of CTAB by UV spectra without probe[J]. Acta Chimica Sinica, 2006, 64(24): 2437-2441. DOI:10.3321/j.issn:0567-7351.2006.24.009 |