 2019, Vol. 47
2019, Vol. 47文章信息
- 刘峣, 武玉琳, 屈云鹏, 郭文英, 梁丽萍
- LIU Yao, WU Yu-lin, QU Yun-peng, GUO Wen-ying, LIANG Li-ping
- Ba2+在KDP(100)表面吸附的密度泛函理论研究
- Density functional theory study of Ba2+ adsorption on KDP (100) surface
- 材料工程, 2019, 47(11): 123-127
- Journal of Materials Engineering, 2019, 47(11): 123-127.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2018.001028
-
文章历史
- 收稿日期: 2018-08-29
- 修订日期: 2019-06-04




2. 临沂市科学技术合作与应用研究院, 山东 临沂 276000
2. Linyi Institute of Science and Technology Cooperation and Application, Linyi 276000, Shandong, China
磷酸二氢钾(KH2PO4,KDP)晶体是一种综合性能优异的非线性光学材料,其非线性光学系数d36多年来一直被作为晶体非线性性质的参比标准。由于KDP晶体具有透过波段宽(近紫外~近红外)、电光系数和非线性光学系数大、损伤阈值高(大于15J/cm2,1ns)、晶体生长尺寸大(直径500~600mm)等特点[1-4],在激光变频、电光调制以及光快速开关等领域一直有着不可替代的地位,是大功率激光系统的首选材料[5]。
为了提高KDP晶体的生长质量和光学性能,人们对KDP晶体做了广泛而深入的研究。然而,在KDP晶体生长过程中,试剂原料和母液不可避免地会混入各种杂质,严重影响着晶体的生长习性和光学稳定性。KDP晶体中的杂质主要包括3种:无机杂质、有机物以及金属离子等[6-8]。其中,具有较强氢键结合能力的无机杂质,如磷酸盐等,容易吸附在晶体的(101)面上,阻碍晶体的生长[9]。细菌、微生物以及各种有机添加剂等,对晶体的激光损伤阈值也有着较大的影响[10]。金属离子是KDP晶体中最常见的杂质,即使是非常微量的金属杂质,也会对KDP晶体的生长过程和光学性质产生明显的影响。研究表明,金属离子容易吸附在KDP晶体的(100)表面,抑制柱面的生长,使晶体变得细长[11-13]。另外,金属颗粒的存在会增加KDP晶体中的光散射,使激光在紫外波段透过率降低,最终造成晶体的激光损伤阈值明显降低[14]。
Ba2+是KDP原材料中常见的一种杂质离子。实验发现,Ba2+同其他金属离子类似,易吸附在KDP晶体的(100)表面上,对晶体的生长有着明显的抑制作用[15]。随着生长溶液中Ba2+含量的增加,KDP晶体对紫外波段的光吸收会明显增加,同时,当Ba2+的质量分数达到一定程度时,晶体会产生开裂的现象[16]。由此可见,Ba2+对KDP晶体的影响不容忽视。虽然已经对此进行了一定的研究,但Ba2+在KDP表面的吸附机理尚不明确。本工作通过密度泛函理论(DFT)研究了Ba2+在KDP晶体(100)表面的吸附行为,对表面结构、吸附能量、电子转移情况等进行了详细分析,探讨了Ba2+与KDP晶体表面之间相互作用的本质。
1 计算方法KDP晶体属于四方晶系,其理想外形由4个柱面和8个锥面,即4个(100)面和8个(101)面构成[17]。金属离子易吸附在KDP晶体的柱面上,因此选取(100)表面作为吸附基底。表面模型由包含96个原子的3层1×1(x,y方向)的面层组成,z方向与所切的表面相垂直。为了避免相邻表面之间的相互影响,在各表面之间设置3nm厚度的真空层。在结构优化过程中,Ba2+和KDP晶体(100)基底的所有原子充分地弛豫,以探索最稳定的吸附构型。
研究方法为基于密度泛函理论(density functional theory, DFT)[18]的第一性原理计算方法,所有的计算均运用Material Studio 8.0中的CASTEP(cambridge sequential total energy package, CASTEP)程序完成。采用广义梯度近似GGA(generalized gradient approximation, GGA)下的PBE(perdew-burke-ernzerhof, PBE)[19]泛函来描述交换关联作用,使用超软赝势(ultrasoft pseudopotential, USPP)[20]描述离子实与价电子的相互作用,设置H(1s1),O(2s2, 2p4),P(3s2, 3p3),K(3s2, 3p6, 4s1),Ba(5s2, 5p6, 6s2)为价态电子。通过总能量收敛性测试,几何优化及电子结构计算的平面波截止能设置为420eV。布里渊区的k点取样设置为2×2×l。模型的结构优化采用BFGS(broyden-fletcher-goldfarb-shanno, BFGS)算法,优化参数设置如下:原子间相互作用力的收敛标准为0.5eV/nm;能量收敛标准设置为2×10-5eV/atom;晶体内部应力的收敛标准为0.1GPa;原子最大位移收敛标准为0.0002nm。
2 结果与分析 2.1 吸附能与吸附构型图 1为Ba2+在KDP晶体(100)表面的初始吸附位置,图 1中白色圆球为H原子,红色为O原子,淡紫色为P原子,深紫色为K原子。为便于观察所选择的吸附位置,图 1仅显示了KDP(100)表面最顶层的磷酸根基团及钾原子,其余原子未全部显示,图中尺寸较大的原子代表靠近表面的原子,较小的原子代表离表面较远的原子。由于金属离子易与KDP晶体(100)表面裸露的磷酸根基团产生相互作用[21],因此本研究主要选取表面H,O原子周围的位点作为初始吸附位置,包括:H-O桥位(B, D, J),O-O桥位(F, L),O顶位(A, E, G),H顶位(C),具体位置如图 1所示。通过对Ba2+在不同位置的吸附过程进行模拟,得到了不同的吸附构型与吸附能。计算吸附能所用的公式如下:
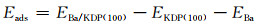
|
(1) |

|
图 1 Ba2+在KDP(100)表面的初始吸附位置 Fig. 1 Initial adsorption positions of Ba2+ on the KDP (100) surface |
式中:Eads为Ba2+在KDP(100)表面的吸附能;EBa/KDP(100)为Ba2+在KDP(100)表面吸附后整个体系的能量;EKDP(100)和EBa分别为清洁(100)表面和单个Ba2+的能量。利用吸附能作为Ba2+在KDP晶体(100)表面吸附能力强弱的判据,能量越低,意味着吸附体系越稳定。
表 1为Ba2+在KDP晶体(100)表面不同位置的吸附能及对应的吸附构型。由表 1中数据可见,吸附能处于-0.80~-1.14eV的范围内,数值均为负值,意味着Ba2+在KDP(100)表面的吸附为自发进行的放热反应。在结构优化的过程中,Ba2+的位置发生了不同程度的移动,最终形成了3种较为稳定的吸附构型,分别用Ⅰ,Ⅱ,Ⅲ表示。图 2为3种吸附构型的俯视图,图 2中灰色圆球代表Ba2+。从表 1及图 2(a)可以看出,对于吸附构型Ⅰ,初始位置分别为A, B, C,Ba2+在结构优化时逐渐移动到了O原子的上方。其中B位置的吸附相对更稳定,对应的吸附能大小为-0.83eV。对于吸附构型Ⅱ,初始位置为D, E, F, G, J的Ba2+,在吸附过程中逐渐移动到了O—O桥位附近(见图 2(b))。其中,当Ba2+的初始位置为E和F时,吸附能均为-1.14eV,吸附能最低,意味着E和F位置的吸附是所有吸附中最稳定的。在吸附构型Ⅲ中,L位置的吸附能为-1.07 eV,吸附前后Ba2+的位置未发生明显的变化,稳定在两个O原子之间(见图 2(c))。需要注意的是,吸附构型Ⅱ和构型Ⅲ中Ba2+最终位置均为O—O桥位,但不同之处在于构型Ⅱ中两个O原子来自同一个磷酸根基团,而Ⅲ中的O原子则由两个不同的磷酸根贡献,前者的吸附构型相对更稳定。
| Position | A | B | C | D | E | F | G | J | L |
| Eads /eV | -0.80 | -0.83 | -0.82 | -1.09 | -1.14 | -1.14 | -1.13 | -1.12 | -1.07 |
| Configuration | Ⅰ | Ⅰ | Ⅰ | Ⅱ | Ⅱ | Ⅱ | Ⅱ | Ⅱ | Ⅲ |

|
图 2 Ba2+在KDP(100)表面的3种吸附构型 (a)Ⅰ; (b)Ⅱ; (c)Ⅲ Fig. 2 Three adsorption configurations of Ba2+ on the KDP (100) surface (a)Ⅰ; (b)Ⅱ; (c)Ⅲ |
在之前的工作中[21],发现金属离子与KDP晶体(100)表面的氧原子及最外层的氢原子H1(具体原子编号见图 2(b))有一定的相互作用,因此在本工作中计算了Ba2+与表面H,O原子的距离。由图 2中给出的距离(单位为nm)大小可知,在3种吸附构型中,Ba2+与其周围O原子的间距在0.254~0.274nm之间,与H1原子的距离在0.323~0.353nm之间。可以看出,虽然吸附位置不同,但3种构型中相应的原子间距较为接近,意味着此距离为Ba2+与KDP(100)表面相互作用的稳定距离。同时,为了确定Ba2+吸附对表面结构的影响,对比了吸附前后KDP晶体(100)表面化学键的长度变化。表 2为Ba2+吸附前后KDP(100)表面的键长参数,由表中数据可见,与吸附之前相比,表面的P—O键、H—O键以及K—O键的长度都有不同程度的变化,大部分的化学键被拉长,同时也有部分化学键的长度有轻微的缩短现象。值得注意的是,在最稳定的吸附构型Ⅱ中,表面氢原子H2与氧原子O3之间的距离由吸附前的0.108nm变为0.144nm,变化非常明显。与其对应的氢键的形式和结构也发生了变化,由O1…H2-O3变为了O1-H2…O3(见图 2(b))。在吸附构型Ⅲ中,靠近表面内部的一个氢键结构也发生了明显的变化,H—O以及H…O键的长度由0.107,0.139nm分别变为0.116,0.126nm。化学键的变化意味着Ba2+吸附对表面结构产生了一定的影响,从而进一步影响着KDP晶体的生长过程。
| Bond | Bond length/nm | |||
| Before adsorption | Ⅰ | Ⅱ | Ⅲ | |
| P1-O1 | 0.151 | 0.154 | 0.155 | 0.152 |
| P2-O2 | 0.148 | 0.148 | 0.151 | 0.150 |
| P2-O3 | 0.155 | 0.156 | 0.153 | 0.156 |
| H1-O4 | 0.098 | 0.101 | 0.101 | 0.103 |
| H2-O3 | 0.108 | 0.102 | 0.144 | 0.103 |
| K1-O1 | 0.281 | 0.290 | 0.283 | 0.294 |
| K1-O2 | 0.269 | 0.267 | 0.276 | 0.291 |
| K2-O2 | 0.277 | 0.274 | 0.279 | 0.290 |
为了探究Ba2+与KDP晶体(100)表面相互作用的本质,计算了差分电子密度。差分电子密度Δρ的计算公式如下:

|
(2) |
式中:ρBa/KDP(100),ρBa,ρKDP(100)分别代表吸附体系、孤立Ba2+和清洁KDP(100)表面的电子密度。图 3为最稳定的吸附构型Ⅱ中Ba2+与KDP晶体(100)表面的差分电子密度,黄色和蓝色分别代表电子密度的增加和减少,等值面设置为±20e/nm3。可以看出,Ba2+在KDP(100)表面吸附之后,电子密度发生了明显的变化。在靠近O2和O3原子顶部的位置,以及Ba2+和H1原子之间的位置,电子密度明显增加。不同的是,Ba2+与O2,O3原子之间的电子密度变化主要集中在单个原子周围,表明它们之间可能存在少量电子的转移,Ba2+和O原子之间存在着离子键的作用。Ba2+和H1原子之间的位置出现了电子密度的增加,意味着它们之间可能存在一定的共价相互作用。

|
图 3 Ba2+在KDP(100)表面最稳定吸附构型的差分电子密度 (a)沿z轴方向的俯视图; (b)侧视图 Fig. 3 Difference electron density for the most stable adsorption configuration of Ba2+ on the KDP (100) surface (a)top view along the z-direction; (b)side view |
吸附前后电子密度的变化必然伴随着电荷转移,通过对Hirshfeld电荷转移情况进行分析,发现吸附之后Ba2+失去了0.19个电子,而O2,O3,H1分别得到了0.03,0.03,0.09个电子。也就是说,在吸附过程中Ba2+向KDP(100)表面转移了部分电子,而这些电子主要提供给了O2,O3,H1等原子。需要注意的是,H1原子从Ba2+处得到电子,同时图 3中电子密度增加的区域偏向于H1原子,这意味着Ba2+和H1原子之间的共价相互作用具有一定的极性特征。
为了进一步确定Ba2+与KDP晶体(100)表面的相互作用机理,对化学键重叠布居数[22]进行了计算,吸附构型Ⅱ中Ba-O2和Ba-O3的重叠布居数均为负值(-0.38,-0.46),意味着离子键的形成。Ba-H1的重叠布居数为正值(0.14),代表它们之间存在较弱的共价作用。化学键重叠布居的计算结果与差分电子密度及Hirshfeld电荷转移的分析吻合一致。另外,对于吸附构型Ⅰ和吸附构型Ⅲ,它们与构型Ⅱ具有相似的Ba—O,Ba—H原子间距(见图 2),可推测3种吸附构型中Ba2+与KDP(100)表面具有相同的作用机制,即Ba2+与O原子之间的结合方式同样为离子键,而Ba2+与H1之间则存在一定的共价作用。
3 结论(1) Ba2+在KDP晶体(100)表面不同位置的吸附能均为负值,大小为-0.80~-1.14eV,意味着Ba2+可以自发吸附到KDP(100)表面上,从而进一步对KDP晶体的生长过程和光学性能产生影响。
(2) Ba2+在KDP晶体(100)表面共形成3种稳定的吸附构型,最佳吸附位置分别为O原子顶位、来自同一磷酸根基团的两个O原子间的桥位,以及来自不同磷酸根基团的两个O原子间的桥位,其中第二种吸附构型最稳定。在3种构型中,Ba2+与表面O原子均通过离子键结合,与表面最外层的H原子产生一定的共价相互作用。
(3) Ba2+对KDP晶体(100)表面结构有一定的影响,表面的P—O键、H—O键以及K—O键均有不同程度的伸长或缩短,同时表面的氢键结构和形式也发生了明显的变化。
| [1] | ZAITSEVA N, CARMAN L. Rapid growth of KDP-type crystals[J]. Progress in Crystal Growth & Characterization of Materials, 2001, 43 (1): 1–118. |
| [2] | ZAITSEVA N P, RASHKOVICH L N, BOGATYREVA S V. Stability of KH2PO4, and K(H, D)2PO4, solutions at fast crystal growth rates[J]. Journal of Crystal Growth, 1995, 148 (3): 276–282. DOI: 10.1016/0022-0248(94)00606-7 |
| [3] | CRAXTON R S, JACOBS S D, RIZZO J, et al. Basic properties of KDP related to the frequency conversion of 1μm laser radiation[J]. Journal of Quantum Electronics IEEE, 1981, 17 (9): 1782–1786. DOI: 10.1109/JQE.1981.1071349 |
| [4] |
张磊.水分子在KDP晶体(100)和(101)面上吸附行为的密度泛函理论研究[D].济南: 山东大学, 2017. ZHANG L. DFT study of water molecule adsorption on the KH2PO4 (100) and (101) surfaces[D]. Jinan: Shandong University, 2017. http://cdmd.cnki.com.cn/Article/CDMD-10422-1017070372.htm |
| [5] |
朱胜军, 王圣来, 丁建旭, 等. 过饱和度对KDP晶体生长与光学性能的影响研究[J].
人工晶体学报, 2013, 42 (10): 1973–1977.
ZHU S J, WANG S L, DING J X, et al. Effect of supersaturation on growth and optical properties of KDP crystal[J]. Journal of Synthetic Crystals, 2013, 42 (10): 1973–1977. DOI: 10.3969/j.issn.1000-985X.2013.10.001 |
| [6] | ZAITSEVA N, CARMAN L, SMOLSKY I, et al. The effect of impurities and supersaturation on the rapid growth of KDP crystals[J]. Journal of Crystal Growth, 1999, 204 (4): 512–524. DOI: 10.1016/S0022-0248(99)00194-3 |
| [7] | BELOUET C, DUNIA E, PÉTROFF J F. X-ray topographic study of defects in KH2PO4, single crystals and their relation with impurity segregation[J]. Journal of Crystal Growth, 1974, 23 (4): 243–252. DOI: 10.1016/0022-0248(74)90065-7 |
| [8] | OWCZAREK I, SANGWAL K. Effect of impurities on the growth of KDP crystals:mechanism of adsorption on (101) faces[J]. Journal of Crystal Growth, 1990, 102 (3): 574–580. DOI: 10.1016/0022-0248(90)90416-I |
| [9] | FU Y J, GAO Z S, LIU J M, et al. The effects of anionic impurities on the growth habit and optical properties of KDP[J]. Journal of Crystal Growth, 1999, 198 (3): 682–686. |
| [10] | YOKOTANI A, NISHIDA Y, FUJIOKA K, et al. A chromogenic limulus test for detection of microbes that decreases the laser damage threshold of potassium dihydrogen phosphate crystals[J]. Journal of Applied Physics, 1987, 61 (9): 4696–4698. DOI: 10.1063/1.338384 |
| [11] |
丁建旭, 刘冰, 王圣来, 等. Cr3+掺杂对快速生长KDP晶体的生长习性和光学性能的影响[J].
无机材料学报, 2011, 26 (4): 354–358.
DING J X, LIU B, WANG S L, et al. Effect of Cr3+ dopant on growth habit and optical properties of rapid grown KDP crystal[J]. Journal of Inorganic Materials, 2011, 26 (4): 354–358. |
| [12] | RASHKOVICH L N, KRONSKY N V. Influence of Fe3+, and Al3+, ions on the kinetics of steps on the {100} faces of KDP[J]. Journal of Crystal Growth, 1997, 182 (3/4): 434–441. |
| [13] | EREMINA T A, KUZNETSOV V A, EREMIN N N, et al. On the mechanism of impurity influence on growth kinetics and surface morphology of KDP crystals-Ⅱ:experimental study of influence of bivalent and trivalent impurity ions on growth kinetics and surface morphology of KDP crystals[J]. Journal of Crystal Growth, 2005, 273 (3/4): 586–593. |
| [14] | HOPPER R W, UHLMANN D R. Mechanism of inclusion damage in laser glass[J]. Journal of Applied Physics, 1970, 41 (10): 4023–4037. DOI: 10.1063/1.1658407 |
| [15] | EFREMOVA E P, SUKHANOVSKAYA A I, KUZNETSOV V A. Effective distribution coefficients of cation impurities in KDP crystals[J]. Inorganic Materials, 2004, 40 (6): 636–640. DOI: 10.1023/B:INMA.0000031999.11469.89 |
| [16] | ZHENG G, SU G, ZHUANG X, et al. Growth and properties of Ba-doped KDP crystals[J]. Crystal Research & Technology, 2010, 43 (8): 811–816. |
| [17] | MOROSIN B, SAMARA G A. Pressure effects on the lattice parameters and structure of KH2PO4-type crystals[J]. Ferroelectrics, 1972, 3 (1): 49–56. DOI: 10.1080/00150197108237683 |
| [18] | DELLEY B. An all-electron numerical method for solving the local density functional for polyatomic molecules[J]. Journal of Chemical Physics, 1990, 92 (1): 508–517. DOI: 10.1063/1.458452 |
| [19] | PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77 (18): 3865–3868. DOI: 10.1103/PhysRevLett.77.3865 |
| [20] | VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review:B, 1990, 41 (11): 7892. DOI: 10.1103/PhysRevB.41.7892 |
| [21] | WU Y L, ZHANG L, LIU Y, et al. Adsorption mechanisms of metal ions on the potassium dihydrogen phosphate (100) surface:a density functional theory-based investigation[J]. Journal of Colloid & Interface Science, 2018, 522 : 256–263. |
| [22] | MULLIKEN R S. Electronic Population analysis on LCAO-MO molecular wave functions[J]. Journal of Chemical Physics, 1955, 23 (10): 1841–1846. DOI: 10.1063/1.1740589 |