 2019, Vol. 47
2019, Vol. 47文章信息
- 刘香军, 杨吉春, 贾桂霄, 杨昌桥, 蔡长焜
- LIU Xiang-jun, YANG Ji-chun, JIA Gui-xiao, YANG Chang-qiao, CAI Chang-kun
- 金属元素掺杂α-Fe(N)体系的电子结构及力学性能的第一性原理计算
- Electronic structures and mechanical properties of metal doped α-Fe (N): a first principle calculation
- 材料工程, 2019, 47(9): 72-77
- Journal of Materials Engineering, 2019, 47(9): 72-77.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2018.000391
-
文章历史
- 收稿日期: 2018-04-10
- 修订日期: 2019-03-11




2. 内蒙古科技大学 工业技术研究院, 内蒙古 包头 014010
2. Institute of Industrial Technology Research, Inner Mongolia University of Science and Technology, Baotou 014010, Inner Mongolia, China
N对钢材性能有着显著的影响,如强度、硬度均随N含量的增加而提高,韧性却不降低[1]。同时N还能提升高氮钢的抗摩擦、抗腐蚀能力[2-4]。由于常压下N在钢液中的溶解度很低[5],通常采用高压充氮[6]和添加固溶金属氮化物的方法[7-9]来提高N在钢中的溶解度。但高压充氮对设备的要求高、成本大,因此添加固溶金属氮化物(如Cr, Mn, Mo和V等的氮化物)来提高钢中氮含量是一种经济合理的有效方法。
在实际研究中,要实现氮在钢液中固溶量的精确测定以及分布稳定性的控制较为困难。此外,由于实验条件的局限性,尚不能够精确表征N在高氮钢中的分布以及合金元素对N的影响规律。同时,固氮金属原子对氮和铁的相互作用机制及其对力学性能的影响鲜见报道。
基于密度泛函理论的第一性原理能够不受实验条件的局限性准确预测合金的电子结构与热力学信息,相比实验研究能更准确地反映材料的本征物性。研究发现[10-14],通过第一性原理计算可以得到掺杂金属或C, N等小原子对不同结构Fe稳定性的影响,并可较为准确地探究不同原子在基体中的占位倾向,深入探索掺杂原子对基体稳定性的影响以及作用机理。
因此,本工作以高氮钢为研究对象,采用第一性原理计算掺杂金属元素对α-Fe(N)稳定性的影响规律;以元素周期表中位于Fe同一周期附近的6种金属Ti, V, Cr, Mn, Co和Ni为掺杂元素,探究金属氮化物在钢中固溶的稳定性、体系电子结构以及力学性能,研究具有不同半径和电子结构的掺杂元素在α-Fe(N)中的稳定规律。
1 计算模型与方法本研究所有计算采用广义梯度近似(generalized gradient approximation, GGA)[15]中的PBE (Perdew-Burke-Ernzerh)[16]泛函方法,选取450eV的平面波截止能和4×4×4的MP-k(Monhkorst-Pack)[17-18]网格大小。自洽循环能量收敛设为1.0×10-4eV/atom,力收敛为0.002eV/nm。本工作研究了α-Fe(N)-M体系的稳定性、电子结构和弹性常数,所有计算由VASP程序完成[19]。
α-Fe晶胞具有体心立方结构,采用GGA-PBE方法获得的晶格常数为0.283nm,与实验结果0.287nm[20]及理论计算值0.282nm[21]几乎一致。选取2×2×2大小的超胞,见图 1。在体心立方α-Fe中,N原子溶入八面体间隙所受到的阻力比溶入四面体间隙小,N易固溶在八面体间隙位置[22-24],因此在本工作中,研究M(M=Ti, V, Cr, Mn, Co, Ni)原子在N原子固溶在八面体间隙的晶体结构模型中的占位情况。根据M原子与N原子的位置关系,本研究给出3种结构模型:M置换Fe1,其与N最邻近,即体心位置(BN);M置换Fe2,其与N次邻近,即顶角位置(TN);M置换Fe3,其与N不相邻(标识为NN),见图 1。

|
图 1 合金原子M取代α-Fe(N)中的BN, TN与NN位置结构 Fig. 1 Structure of M respectively doped in BN, TN and NN site of α-Fe(N) |
为了描述金属氮化物在α-Fe中稳定情况,使用原子间结合能来表示,计算公式[25]如下:
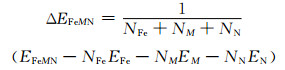
|
(1) |
式中:ΔΕFeMN为晶体的结合能;ΕFeMN为晶体的总能量;ΕFe, ΕM和ΕN分别为Fe, M和N孤立原子的能量,通过将其放置在1nm×1nm×1nm大小的晶胞格子中获得;NFe, NM和NN分别为单胞中Fe, M和N原子的数量。结合能越小,原子间的结合力越强,将需要更高的能量才能使键断裂,晶胞越稳定。
对晶胞结构进行优化后,晶格常数c略大于a,因此计算掺杂结构的力学性能使用满足四方晶系的相应计算公式。根据四方晶系力学稳定性判断条件:对于四方体系,独立弹性常数C11, C12, C13, C33, C44和C66必须同时满足公式(2),晶体才能稳定存在。
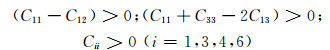
|
(2) |
根据计算出的独立弹性常数,通过Voigt-Reuss-Hill[26]近似,可以进一步计算出各固溶体的体积模量B、剪切模量G和弹性模量E,计算公式见式(3)~(10)[27]:

|
(3) |
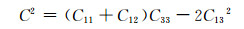
|
(4) |
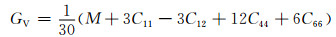
|
(5) |

|
(6) |

|
(7) |
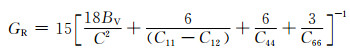
|
(8) |
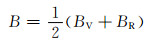
|
(9) |
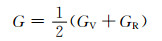
|
(10) |
式中:BV和GV分别为Voigt近似下的体积模量与剪切模量;BR和GR分别为Reuss近似下的体积模量与剪切模量。
2 结果与讨论 2.1 几何结构对掺杂M(M=Ti, V, Cr, Mn, Co, Ni)原子的α-Fe(N)体系进行结构优化,结果表明,体系的晶胞参数均发生了变化,见表 1。本工作只给出各体系3种掺杂结构中具有能量最低体系的几何结构参数。从表 1可以看出,掺杂体系ΔV均增大,且不同掺杂元素对体系晶胞参数变化的影响不同。与α-Fe(N)体系相比,对比Fe原子半径大的掺杂原子V, Cr和Mn,掺杂体系的ΔV几乎不变或者变小,这说明掺杂的V, Cr和Mn与相邻原子之间产生了较强的相互作用。对原子半径小于Fe的Co和Ni的掺杂体系,ΔV稍微变小,这说明掺杂的Co和Ni与相邻原子之间相互作用较弱。
| System | dFe1—N/nm | dFe4—N/nm | dM—N/nm | Lattice parameter | ΔV/% | r/nm | |
| a/nm | c/nm | ||||||
| α-Fe | - | - | - | 0.5662 | 0.5662 | 0 | 0.1720 |
| α-Fe(N) | 0.1704(0.1920)* | 0.1962(0.1920)* | - | 0.5647 | 0.6063 | 6.5100 | - |
| α-Fe(N)-Ti(TN) | 0.1713 | 0.1977 | 0.2095(0.2070)* | 0.5681 | 0.6064 | 7.8300 | 0.2000 |
| α-Fe(N)-V(TN) | 0.1714 | 0.1967 | 0.2034(0.1970)* | 0.5657 | 0.6057 | 6.5300 | 0.1920 |
| α-Fe(N)-Cr(BN) | 0.1708 | 0.1968 | 0.1720(0.1930)* | 0.5634 | 0.6092 | 6.5200 | 0.1850 |
| α-Fe(N)-Mn(BN) | 0.1714 | 0.1967 | 0.2033(0.1920)* | 0.5646 | 0.6040 | 6.0600 | 0.1790 |
| α-Fe(N)-Co(NN) | 0.1708 | 0.1964 | - | 0.5646 | 0.6059 | 6.4200 | 0.1670 |
| α-Fe(N)-Ni(NN) | 0.1708 | 0.1964 | - | 0.5637 | 0.6081 | 6.4400 | 0.1620 |
| * The sum of covalent radius of Fe and N atoms or M and N atoms is given in the bracket. | |||||||
由表 1可见,晶胞中的晶格参数发生了变化,a=b≠c,a与c均不同程度地增大,且c略大于a,晶胞发生了畸变。究其原因是N掺杂在八面体空隙后,N所在八面体的Fe原子位置发生了相应的移动。α-Fe的八面体间隙的半径为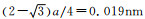
为了研究这些金属原子在α-Fe(N)中的稳定性,本工作计算了M(M=Ti, V, Cr, Mn, Co, Ni)取代掺杂α-Fe(N)的3种可能结构的总能量。为进一步说明M取代掺杂体系的稳定性,本工作计算了3种取代掺杂中最稳定α-Fe(N)-M体系的结合能,并与α-Fe(N)体系进行比较,见表 2。从表 2可以看出,固溶合金原子改变了晶胞的结合能,其中,Ti和V的固溶减小了晶胞的结合能,这表明这些掺杂原子提高了体系稳定性;Cr, Mn和Ni的固溶稍微增大了晶胞的结合能,这表明这些掺杂原子降低了体系的稳定性;Co的固溶不影响晶胞结合能。从表 2还可以看出,由于原子半径的不同,结合能有所差别,对位于与Fe同一周期的左侧金属Ti, V, Cr和Mn及位于与Fe同一周期的右侧金属Co和Ni,结合能的绝对值均随原子半径的减小而减小,且顺序为Ti>V>Cr>Mn和Co>Ni。
| System | E/(eV·atom-1) | Left peak value/eV | Right peak value/eV | Pseudogap/eV |
| α-Fe(N) | -5.207 | -1.051 | 1.338 | 2.389 |
| α-Fe(N)-Ti | -5.290 | -1.053 | 1.812 | 2.865 |
| α-Fe(N)-V | -5.253 | -1.126 | 1.149 | 2.275 |
| α-Fe(N)-Cr | -5.137 | -1.144 | 0.624 | 1.768 |
| α-Fe(N)-Mn | -5.122 | -1.042 | 0.574 | 1.616 |
| α-Fe(N)-Co | -5.207 | -1.105 | 0.757 | 1.862 |
| α-Fe(N)-Ni | -5.176 | -1.139 | 0.549 | 1.687 |
为了揭示α-Fe(N)-M体系中原子间的相互作用情况,本工作计算了最稳定体系的态密度(density of states, DOS)和分波态密度(partial density of states, PDOS)与赝能隙,并探讨了M对α-Fe(N)电子结构的影响规律,见图 2(图 2中虚线表示费米能级)。

|
图 2 Fe16, Fe16N体系(a)和α-Fe(N)-M体系(b)的态密度图 Fig. 2 DOS of Fe, Fe16N(a) and α-Fe(N)-M(b) systems |
纯α-Fe在-54.5~-50.5eV的态来自于Fe3p电子,在-8.0~9.0eV的态来自于Fe3p3d4s的杂化电子,见图 2(a)。掺杂N原子后,与N最邻近的Fe1在-17.0~-16.5eV能量区间产生来自N2s电子的新态,与Fe2及离N较远的Fe3相比,邻近N的Fe1在费米能级附近能量发生明显的离域化,且能量分布从原来的-5.0~3.1eV离域到-8.0~3.1eV,这表明N与Fe1原子在此能量区间发生了较强的相互作用,如图 2(a)所示。掺杂金属原子Ti, V, Cr, Mn, Co和Ni后,与α-Fe(N)体系相比,α-Fe(N)-M体系除了来自由掺杂金属M引入的低能级杂质态,主要在-8.0~6.0eV能量区间有来自M3d和N2p电子的相互作用态,如图 2(b)所示。
图 2中费米能级两侧分别有两个尖峰,且尖峰之间的态密度不为零,这两个尖峰间的距离称为赝能隙,赝能隙越宽,共价性越强,因此赝能隙的大小可用于分析体系共价性的强弱。α-Fe(N)-M体系赝能隙见表 2,从表 2可以看出,掺杂原子M中仅Ti增强了体系的共价性,其他α-Fe(N)-M体系的共价性均被减弱。对位于与Fe同一周期的左侧金属Ti, V, Cr和Mn及位于与Fe同一周期的右侧金属Co和Ni,赝能隙均随原子半径的减小而减小,顺序为Ti>V>Cr>Mn和Co>Ni,这两个顺序规律以Fe为临界,类似于结合能规律。
2.4 力学性能对于各种掺杂体系,计算得到的α-Fe(N)-M体系C11, C12, C13, C33, C44和C66代入公式(3)~(10)分别获得各体系体积模量B、剪切模量G、弹性模量E及体现材料延展性能的B/G力学常数,见表 3。
| System | C11 | C12 | C13 | C33 | C44 | C66 | B/GPa | G/GPa | E/GPa | B/G |
| α-Fe | - | - | - | - | - | - | 183.47 | 77.20 | 203.11 | 2.38 |
| α-Fe(N) | 269.86 | 135.22 | 131.33 | 263.09 | 103.48 | 98.72 | 177.59 | 86.37 | 222.96 | 2.06 |
| α-Fe(N)-Ti | 263.90 | 136.57 | 133.23 | 266.70 | 97.99 | 95.88 | 177.84 | 82.88 | 215.20 | 2.15 |
| α-Fe(N)-V | 268.82 | 134.28 | 135.41 | 271.13 | 103.97 | 97.40 | 179.88 | 86.20 | 222.97 | 2.09 |
| α-Fe(N)-Mn | 264.17 | 128.83 | 132.73 | 265.80 | 100.07 | 97.49 | 175.85 | 84.57 | 218.67 | 2.08 |
| α-Fe(N)-Co | 264.94 | 147.78 | 139.75 | 273.51 | 103.97 | 101.55 | 184.21 | 84.38 | 219.62 | 2.18 |
| α-Fe(N)-Ni | 258.50 | 141.10 | 136.52 | 273.35 | 103.91 | 101.49 | 179.88 | 84.33 | 218.80 | 2.13 |
从表 3可以看出,α-Fe(N)-M体系与纯α-Fe体系相比,剪切模量G和弹性模量E均得到提高,且分别提高了7.4%~11.7%和6.0%~9.8%,这说明与纯α-Fe体系相比,金属氮化物的掺杂提高了体系的刚度及耐磨性,这与掺杂金属元素和N之间形成很强化学键有关。与α-Fe(N)体系相比,剪切模量G和弹性模量E保持不变或略微减小,这说明与α-Fe(N)体系相比,金属氮化物的掺杂使体系的变形能力(剪切模量G)变差,材料的刚性下降,说明加入金属氮化物冶炼的高氮钢硬度低于充氮气冶炼的高氮钢。与纯α-Fe体系相比,除α-Fe(N)-Co体系外,体积模量B均不同程度地减小,但其下降幅度很小,仅为1.9%~4.1%。这表明,掺杂金属Co原子后,体系的不可压缩性最好。对体现材料延展性能的B/G常数进行了分析,与纯α-Fe体系相比,所有掺杂体系均不同程度地减小,减小了8.4%~14.3%,这表明金属氮化物的掺杂降低了材料的韧性。与α-Fe(N)体系相比,由于合金元素的掺杂使B/G不同程度地增大,这说明加入金属氮化物能够不同程度地提高高氮钢的韧性。从表 3还可以看出,除了α-Fe(N)-Co(Co与N不相邻,Co不是有效的固氮元素)以外,掺杂金属原子V后体系的弹性模量E、剪切模量G和体积模量B均最大,这说明常压冶炼高氮钢时,添加V是提高钢材力学性能最有效的固氮元素。
3 结论(1) 位于Fe同一周期左侧的Ti, V, Cr以及Mn与N相邻的位置最稳定,其中,Ti和V优先占据晶胞的顶角位置,Cr和Mn优先占据晶胞的体心位置,位于Fe右侧的Co和Ni与N不相邻时结构最稳定。
(2) Ti与V的掺杂加强了晶胞稳定性,Cr, Mn与Ni的掺杂稍稍削弱晶胞稳定性,Co的掺杂不影响晶胞稳定性。
(3) 与其他α-Fe(N)-M掺杂体系相比,掺杂金属原子V后,体系的弹性模量E、剪切模量G和体积模量B均最大,这说明体系最不容易发生形变,刚性最强。即常压冶炼高氮钢时,添加V是提高钢材力学性能最有效的固氮元素。
| [1] | HARZENMOSER M. Welding of high nitrogen steels[J]. Materials and Manufacturing Processes, 2004, 19 (1): 75–86. DOI: 10.1081/AMP-120027503 |
| [2] | LU L, SHEN Y F, CHEN X H, et al. Ultrahigh strength and high electrical conductivity in copper[J]. Science, 2004, 304 (5669): 422–426. DOI: 10.1126/science.1092905 |
| [3] | GRÄSSEL O, KRÜGER L, FROMMEYER G, et al. High strength Fe-Mn-(Al, Si) TRIP/TWIP steels development-properties-application[J]. International Journal of Plasticity, 2000, 16 (10/11): 1391–1409. |
| [4] | DUSCHER G, CHISHOLM M F, ALBER U, et al. Bismuth-induced embrittlement of copper grain boundaries[J]. Nature Materials, 2004, 3 (9): 621–626. DOI: 10.1038/nmat1191 |
| [5] | SIMMONS J W. Overview:high-nitrogen alloying of stainless steels[J]. Materials Science and Engineering:A, 1996, 207 (2): 159–169. DOI: 10.1016/0921-5093(95)09991-3 |
| [6] | KATADA Y, SAGARA M, KOBAYASHI Y, et al. Fabrication of high strength high nitrogen stainless steel with excellent corrosion resistance and its mechanical properties[J]. Materials and Manufacturing Processes, 2004, 19 (1): 19–30. DOI: 10.1081/AMP-120027495 |
| [7] | BALACHANDRAN G, BHATIA M L, BALLAL N B, et al. Processing nickel free high nitrogen austenitic stainless steels through conventional electroslag remelting process[J]. ISIJ International, 2000, 40 (5): 478–483. DOI: 10.2355/isijinternational.40.478 |
| [8] |
杨吉春, 周莉, 董梦瑶, 等. 增氮降镍对316不锈钢热变形行为的影响[J].
材料热处理学报, 2015, 36 (6): 126–131.
YANG J C, ZHOU L, DONG M Y, et al. Effects of increasing nitrogen and reducing nickel on hot deformation behavior of 316 stainless steel[J]. Transactions of Materials and Heat Treat-ment, 2015, 36 (6): 126–131. |
| [9] |
杨吉春, 张剑, 周莉, 等. V含量对高氮20MnSi钢自然时效性能的影响[J].
特殊钢, 2015, 36 (3): 43–45.
YANG J C, ZHANG J, ZHOU L, et al. Influence of V content on natural ageing properties of high nitrogen steel 20MnSi[J]. Special Steel, 2015, 36 (3): 43–45. DOI: 10.3969/j.issn.1003-8620.2015.03.012 |
| [10] |
陈煜, 姚正军, 张平则, 等. Cr、Mo对DO3-Fe3Al力学性能和电子结构影响的理论研究[J].
稀有金属材料与工程, 2017, 46 (10): 2863–2869.
CHEN Y, YAO Z J, ZHANG P Z, et al. Theoretical research on the effect of Cr, Mo on the mechanical properties and electronic structure of DO3-Fe3Al[J]. Rare Metal Materials and Engineering, 2017, 46 (10): 2863–2869. |
| [11] |
靳磊, 崔向中, 王纯, 等. 钇硅酸盐材料力学性能的第一性原理研究[J].
材料工程, 2017, 45 (7): 48–53.
JIN L, CUI X Z, WANG C, et al. First principle study of mechanical properties of yttrium silicates[J]. Journal of Materials Engineering, 2017, 45 (7): 48–53. |
| [12] |
文平, 李春福, 赵毅, 等. Cr, Mo, Ni在α-Fe(C)中占位、键合性质及合金化效应的第一性原理研究[J].
物理学报, 2014, 63 (19): 197101.
WEN P, LI C F, ZHAO Y, et al. First principles calculation of occupancy, bonding characteristics and alloying effect of Cr, Mo, Ni in bulk α-Fe(C)[J]. Acta Physica Sinica, 2014, 63 (19): 197101. DOI: 10.7498/aps.63.197101 |
| [13] |
王明军, 李春福, 文平, 等. Cr, Mo, Ni在γ-Fe(C)中的键合性质及对相结构稳定性的影响[J].
物理学报, 2016, 65 (3): 037101.
WANG M J, LI C F, WEN P, et al. The bond characters and phase stability effects of Cr Mo and Ni in bulk γ-Fe(C)[J]. Acta Physica Sinica, 2016, 65 (3): 037101. |
| [14] |
张旭昀, 郑冰洁, 郭斌, 等. 高氮奥氏体不锈钢中N与Cr、Mn、Mo键合性质研究[J].
材料导报, 2017, 31 (18): 146–149.
ZHANG X Y, ZHENG B J, GUO B, et al. Theoretical study on bonding characteristics of Cr, Mn, Mo and N in high nitrogen austenitic stainless steel[J]. Materials Review, 2017, 31 (18): 146–149. DOI: 10.11896/j.issn.1005-023X.2017.018.029 |
| [15] | PERDEW J P, CHEVARY J A, VOSKO S H, et al. Atoms, molecules, solids, and surfaces:applications of the generalized gradient approximation for exchange and correlation[J]. Physical Review B, 1992, 46 (11): 6671–6687. DOI: 10.1103/PhysRevB.46.6671 |
| [16] | PERDEW J P, BURKE K, WANG Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system[J]. Physical Review B, 1996, 54 (23): 16533–16539. DOI: 10.1103/PhysRevB.54.16533 |
| [17] | MONKHORST H J, PACK J D. Special points for brillouin-zone integrations[J]. Physical Review B, 1976, 13 (12): 5188–5192. DOI: 10.1103/PhysRevB.13.5188 |
| [18] | PACK J D, MONKHORST H J. "Special points for brillouin-zone integrations"-a reply[J]. Physical Review B, 1977, 16 (4): 1748–1749. DOI: 10.1103/PhysRevB.16.1748 |
| [19] | KRESSE G, HAFNER J. Ab initio molecular dynamics for liquid metals[J]. Physical Review B, 1993, 47 (1): 558–561. DOI: 10.1103/PhysRevB.47.558 |
| [20] | CHAMATI H, PAPANICOLAOU N I, MISHIN Y, et al. Embedded-atom potential for Fe and its application to self-diffusion on Fe(100)[J]. Surface Science, 2006, 600 (9): 1793–1803. DOI: 10.1016/j.susc.2006.02.010 |
| [21] | WANG Y, CURTAROLO S, JIANG C, et al. Ab initio lattice stability in comparison with CALPHAD lattice stability[J]. Calphad, 2004, 28 (1): 79–90. DOI: 10.1016/j.calphad.2004.05.002 |
| [22] |
王笑天.
金属材料学[M]. 北京: 机械工业出版社, 1987: 8-9.
WANG X T. Metal material science[M]. Beijing: Machine Industry Press, 1987: 8-9. |
| [23] |
祝菊生, 王炳洲.
金属理论基础[M]. 北京: 中国宇航出版社, 1992: 32.
ZHU J S, WANG B Z. Theoretical foundation of metal[M]. Beijing: China Astronautic Publishing House, 1992: 32. |
| [24] |
杨正举. 体心立方金属中间隙杂质原子组态的弹性研究:Ⅰ.间隙杂质原子的位置及扩散激活能[J].
物理学报, 1996, 22 (3): 281–293.
YANG Z J. An investigation of the configurations of interstices in B.C.C. metals by elastic method:Ⅰ. the position energy and activation energy of diffusion of interstitial impurities[J]. Acta Physica Sinica, 1996, 22 (3): 281–293. |
| [25] | SAHU B R. Electronic structure and bonding of ultralight LiMg[J]. Materials Science and Engineering:B, 1997, 49 (1): 74–78. DOI: 10.1016/S0921-5107(97)00068-8 |
| [26] | HILL R. The elastic behaviour of a crystalline aggregate[J]. Proceedings of the Physical Society. Section A, 1952, 65 (5): 349–354. DOI: 10.1088/0370-1298/65/5/307 |
| [27] | RESHAK A H, MORTEZA J. DFT calculation for elastic constants of tetragonal structure of crystalline solids with WIEN2k code:a new package (tetra-elastic)[J]. International Journal of Electrochemical Science, 2013, 8 (11): 12252–12263. |