 2019, Vol. 47
2019, Vol. 47文章信息
- 杨宝成, 彭艳, 潘复生, 石宝东
- YANG Bao-cheng, PENG Yan, PAN Fu-sheng, SHI Bao-dong
- 基于分子动力学镁合金塑性变形机制的研究进展
- Research progress in plastic deformation mechanism of Mg alloys based on molecular dynamics
- 材料工程, 2019, 47(8): 40-48
- Journal of Materials Engineering, 2019, 47(8): 40-48.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2017.001301
-
文章历史
- 收稿日期: 2017-10-22
- 修订日期: 2019-04-02



2. 燕山大学 机械工程学院, 河北 秦皇岛 066004;
3. 重庆大学 国家镁合金材料工程技术研究中心, 重庆 400044
2. School of Mechanical Engineering, Yanshan University, Qinhuangdao 066004, Hebei, China;
3. National Engineering Research Center for Magnesium Alloys, Chongqing University, Chongqing 400044, China
镁合金材料具有密度低、比强度高的优点,被广泛应用于汽车和航空航天等领域以实现产品的轻量化[1-3],可以有效地解决日益突出的能源问题和尾气排放等环境问题,备受国内外学者和工程人员的关注。然而,镁合金具有低对称性的密排六方(hexagonal close-packed, HCP)晶体结构,塑性变形时只有较少的滑移系和孪生变形机制开动,易形成较强的基面织构,造成镁合金的力学性能具有明显的各向异性和拉压强度不对称性(strength differential effect, SD)及较低的延展性和成形能力,严重地制约着镁合金的大规模应用[4-7]。因此,研究镁合金的塑性变形机制不仅可以发展和丰富金属塑性变形的基础理论,而且对于改善和提高镁合金的成形能力具有重要的理论意义与工程应用价值。
目前现有的材料表征技术虽然可以有效地观测到材料在微纳米尺度的形貌,但是仍有一些重要的信息无法获取,如:原子运动轨迹、广义层错能和晶界能等。这些信息对于材料的组织、性能调控至关重要。随着计算机技术的发展,计算材料科学成为研究微纳米尺度的重要方法[8-9]。分子动力学(molecular dynamics, MD)方法是近年来计算材料学科发展最快、应用最多的计算方法,已成为金属材料研究其微纳米尺度塑性变形机理的重要方法。本文简要概述了镁合金各种滑移系和孪生等变形机制,总结了分子动力学中HCP结构金属常用的势函数,重点讨论了镁合金微纳米尺度塑性变形机理的研究现状及发展趋势。
1 镁合金塑性变形机制镁合金材料室温下塑性变形能力差,往往表现出强的力学行为各向异性,而强的各向异性本质上是因为不同方向上变形机制在不同条件下有所差异造成的。镁合金中主要的塑性变形机制有晶内滑移、孪生和晶界滑移等,因不同变形机制开启的难易程度不同,故对总塑性变形的贡献也各异。各类变形机制的开动情况也主要受变形温度、合金元素、晶体取向、变形速率和晶粒尺寸等因素影响。
1.1 滑移滑移的本质是位错的运动[10],若依据滑移过程中位错的柏氏矢量的理论可以将镁合金滑移机制划分为〈a〉滑移和〈c+a〉滑移,表 1中列出了不同〈a〉滑移和〈c+a〉滑移所具有的滑移面、滑移方向与独立滑移系数目。需要指出的是:在镁合金晶体中,〈1120〉晶向是最容易发生滑移的方向,〈1123〉晶向是潜在易发生滑移的方向。
| Slip classification | Slip plane | Slip direction | Independent slip system |
| 〈a〉 basal slip system | (0001) | 〈1120〉 | 2 |
| 〈a〉 prismatic slip | {1110}, {1120} | 〈1120〉 | 2 |
| 〈a〉 pyramidal plane slip | {1011} | 〈1120〉 | 4 |
| 〈c+a〉 pyramidal plane slip | {1121}, {1122} | 〈1123〉 | 5 |
温度对镁合金塑性变形能力影响较大。在室温下,镁合金塑性变形机制主要依赖(0001)〈1120〉滑移提供的2个独立的滑移系[12],非基面滑移一般只在应力较集中的晶界附近启动,这是室温变形差的主要原因。随着温度逐渐升高,(0001)滑移系开动的临界分切应力(critical resolved shear stress, CRSS)变化不大,非基面滑移系的CRSS下降很快,当温度高于350℃时,基面和非基面滑移系的CRSS值非常接近[13]。此时,各类滑移系都能顺利开动,提供充足的独立滑移系满足塑性变形的要求,镁合金塑性成形能力得到有效提高。Agnew等[14]在研究AZ31镁合金高温塑性变形时,也发现〈c+a〉非基面滑移开动大幅度提升了塑性成形能力。
此外,改变晶粒尺寸、调控织构分布和添加合金元素等方式均可以影响镁合金的成形能力。从晶界协调变形的角度来说,晶粒细化能够缩短位错滑程[15],晶粒易发生转动,晶粒取向易趋向于利于滑移的方向,晶粒细化还可以激活镁合金中潜在的非基面滑移系,从而有效改善室温塑性变形。镁合金热加工中,易形成平行于挤压方向的基面织构[16],后续再沿挤压方向发生变形时基面滑移就很难开动,材料会沿基面变脆。因此,可以通过调控晶粒转动和定向流动,减少基面织构的生成,以保证高的镁合金变形量。在镁合金材料中,合金元素的添加往往引起晶体结构和合金层错能的改变,进而影响其塑性变形能力。如:添加锌和锂元素能降低c/a值[17],可激活非基面滑移系,使镁合金在较低温度下也具有较好的塑性。
1.2 孪生在镁合金中, 孪生作为另一种晶内塑性变形机制发挥着重要作用。在镁合金实际成形中,孪生与滑移往往互相竞争,通过晶粒转动调节晶体取向并释放晶界应力集中, 从而促使滑移和孪生能够交替进行,使材料获得较大的变形。然而,若是某种孪生的Schmid因子小于零,即孪生切变方向上剪切应力分量方向与孪生切变方向相反,即便外加载荷很大,孪生开动所需的CRSS值较小,此时这类孪生也不可能开启。
材料的受力情况和轴比c/a值的改变都会对孪生产生较大影响。外加载荷的作用方式决定孪生是否发生,而c/a值的改变往往引起原子最密排面和相关晶体学参数的改变。镁合金中常见孪晶模式和孪生要素如表 2所示[18-19],其拉伸孪晶以{1012}和{1121}为主,只有当载荷作用方向沿轴线方向受拉或沿垂直于轴线方向受压时才会发生;压缩孪晶则以{1122}和{1011}为主,往往发生在高温塑性变形中,并且很少以单一形态出现。此外,{1012}〈1011〉孪晶系可使晶粒的c轴趋向平行于压缩轴, 形成基面织构, 导致强的各向异性力学行为[20]。
| K1 | K2 | η1 | η2 | Nt | NS/Nt | q | s | A/(°) |
| {1012} | {1012} | 〈1011〉 | 〈1011〉 | 8 | 3/4 | 9 | 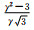
|
86.3 |
| {1011} | {1013} | 〈1012〉 | 〈3032〉 | 32 | 7/8 | 8 | 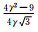
|
56.1 |
| {1022} | {1024} | 
|
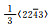
|
12 | 2/3 | 6 | 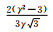
|
63.3 |
| {1021} | {0002} | 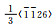
|

|
8 | 1/2 | 2 | 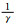
|
38 |
| Note: K1, K2-the first and second undistorted plane; η1, η2-twin directions; Nt-the number of atoms in each twin element; NS-the number of sheared atoms; q-the number of K1 in the twins; s-the twinning shear expressec in terms of the axial ratio; A-the twin orientation angle; γ-the value of c/a | ||||||||
在镁合金变形中,拉伸孪晶主要发生在变形初期,其对应变有贡献[21-22],可以在晶粒中长大并大量增殖;压缩孪晶则主要发生在变形结束阶段,有利于应力松弛[23],始终保持相对细小的形貌。另外,在发生一次孪晶的晶粒内还可能发生二次孪晶,这样二次孪晶和滑移机制之间的交互作用能够带来很大的应变。Barnett等[24-26]结合EBSD技术和仿真模拟方法对AZ31镁合金塑性变形中生成的二次孪晶进行了研究,重点对二次孪晶生成机理做了合理解释。其后,Cizek等[27]利用TEM技术对AZ31镁合金拉伸断口附近的紧缩孪晶进行研究,并验证了Barnett等提出的AZ31镁合金中二次孪晶生成机理。
1.3 晶界滑移在多晶镁合金塑性变形中,除了要考虑晶内塑性变形机制外,也必须要考虑晶间塑性变形机制发挥的重要作用。镁合金中最重要的晶间变形机制是相邻晶粒之间的晶界滑移[11],通常只在高温低应变条件下才能发生。
余琨等[28]在研究镁合金晶粒细化工艺时,发现添加稀土后的AZ31RE镁合金室温拉伸时伸长率都超过了20%,是相同条件下常规镁合金变形的3~4倍,仅仅依靠滑移和孪生滑移系室温拉伸伸长率一般都很低,因此添加稀土后晶界滑移顺利开动起到了重要作用,并通过对比稀土元素添加前后的AZ31RE镁合金的显微组织,解释了晶界滑移的作用机理。此外,温度也对晶界滑移变形机制的影响很大,与滑移变形机制相同,随着温度升高,由晶界滑移引起的应变占总应变的比率逐步增大,特别是在镁合金高温超塑性变形中,晶界滑移起主导作用。郑翊等[29]通过对ZK60镁合金板材超塑性变形前后的组织演化特征和断口形貌进行分析,证实了晶界滑移是主要变形机制,位错蠕变起协调作用。
2 分子动力学方法分子动力学方法是在评估和预测材料组织结构与性能方面,基于微纳米尺度下模拟组成原子和分子的一种重要计算方法。其最大的优点是不受样品制备和测试方法的约束,建立材料微观结构和宏观力学性能之间的联系,从本源解释材料宏观力学行为。其实质是通过计算机模拟由原子或分子所构成的多体体系中微观粒子之间相互作用和运动,获得体系中原子或分子的运动轨迹,再参考物理统计的方法得到物质的结构和宏观性质等[30]。
相比于第一性原理计算方法,由于体系内粒子间的相互作用关系由包含经验或半经验参数的势函数直接给出,分子动力学的计算量相对很小,因此可以模拟粒子数目庞大的体系,已有文献报道可以模拟数以十亿计的原子体系[31]。近年来,将分子动力学方法与第一性原理方法结合的理论和算法取得了重大突破,这使得分子动力学方法可以跳出传统经验势场确定性的限制。可以预见,在材料理论研究和开发新材料中分子动力学会发挥越来越重要的作用。
2.1 分子动力学基本原理分子动力学方法是基于两个假设,其一是全部粒子的运动均满足经典牛顿运动规律,二是粒子之间的相互作用满足叠加原理,显然分子动力学方法是一种近似计算方法。分子动力学基本原理是通过为模拟体系选取合适的势函数、相关系综和特定的边界条件,确定其主要的模拟范畴。对给定的牛顿运动方程,在给定初始条件下,通过数值方法求解(基本上是用有限差分法来对二阶微分方程进行积分求解),在到达给定的收敛条件后获得一个最终的位置坐标,之后对有用计算信息经过后处理方法给出模拟体系的统计量,如热力学、动力学和光学性质等。
2.2 HCP结构金属常用势函数势函数能否精确再现原子间的相互作用将直接决定计算结果的可靠性与准确性,因此,为待模拟体系选择可靠的势函数尤为关键[32]。对于金属体系,所有的电子为体系共用并可以在晶体内运动,人们通过参考密度泛函理论(density functional theory, DFT),将原子周围的环境考虑为局域电子密度,结合宏观的实验结果和理论值,在非金属体系势函数基础上提出了新的半经验势函数模型,现有包括嵌入原子势(embedded atom method, EAM)、修正型嵌入原子势(modified embedded atom method, MEAM)以及多种演化势等可供选择。以下简要介绍常用于HCP结构金属的3种势函数:
(1) Liu-EAM
Liu等[33]通过力匹配和实验数据模拟的方法,开发了适用于HCP结构金属的半经验EAM势函数,记为Liu-EAM。其函数具体形式为:
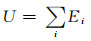
|
(1) |

|
(2) |

|
(3) |
式中:U为体系总势能,即体系中所有原子势能之和;Ei为任意原子i的势能;ϕ(rij)为相距rij的原子i和原子j之间的对势能;F(ρi)为将原子i嵌入背景电荷密度ρi中所需要的能量;ρ(rij)为原子i周围其他原子所贡献的电荷密度。
(2) Sun-EAM
Sun等[34]针对Liu-EAM势在熔点温度方面与实验数据拟合性差等问题,对势函数参数进行优化,提出了新的势函数模型,记为Sun-EAM。该势函数表达式为:

|
(4) |
式中N为该体系中的原子总数。
(3) 第二近邻修正型嵌入原子势
Baskes在EAM势的基础上考虑了角度方向开发了MEAM势[35],但只考虑了第一近邻原子相互作用,不能有效应用于HCP结构合金材料的分子动力学计算。后来,Kim等[36-37]在MEAM势的基础上,进行了参数修正提出了第二近邻修正型嵌入原子势(2NN MEAM),能够很好地表征不同原子间相互作用机理。
总体来说,Liu-EAM势在晶格常数、堆垛层错能和空位形成能等方面与实验数据拟合较一致,但在材料有关热力学性质计算中误差很大。Sun-EAM势通过参数修正可用于有关热力学性质等方面计算,但是在模拟HCP结构合金时与实验结果误差很大。2NN MEAM势考虑了第二近邻原子相互作用,因其精度高和规范性可以满足合金材料的计算要求,现广泛应用于HCP结构合金的模拟中。
3 基于分子动力学的镁合金塑性变形机制研究现状在金属材料塑性变形方面,分子动力学方法是一种有效的计算工具,可以从微纳米尺度解释滑移和孪晶等变形机制的作用机理。在塑性方面,国内外学者现主要集中于BCC结构和FCC结构金属的研究,而对于HCP结构金属材料研究较少。国外基于分子动力学方法对镁合金塑性变形机制的研究起步较早,研究比较成熟;国内则起步较晚、研究相对滞后。总体来说,对镁合金塑性变形机理的研究主要涉及孪晶的形核与长大机理、位错和滑移的作用机理、滑移与孪生之间的交互关系、合金元素和晶体取向等因素对塑性变形的影响。
3.1 基于分子动力学的孪生变形机制孪生是金属材料塑性变形中的一种重要变形机制,其过程包括孪晶形核和孪晶长大[38-39],孪晶生长实质上是孪晶位错在孪晶面上的运动。Wang等[40-41]将分子动力学方法与EBSD实验结合起来,在分子模拟和拓扑学分析基础上提出了镁合金的孪晶形核机理。研究发现,孪晶均匀形核必须要具备极高的应力条件,因而孪晶更加偏向于在晶界附近或应力集中处非均匀形核。
Aghababaei和Joshi[42]应用分子动力学方法研究了镁单晶拉伸孪晶的演化过程。在模拟中,选用了Sun-EAM势函数、等温等压系综(NPT)和周期性边界条件,着重分析了{1012}和{1121}两类拉伸孪晶的演化情况。研究发现:{1121}孪晶系并不依赖于有无初始缺陷存在,而{1012}孪晶在包含空穴或缺陷的晶体中更容易出现;在整个演化过程中,{1121}和{1012}两种孪晶都被激活,但由于{1012}孪晶对应变十分敏感,随着变形增加{1012}孪晶起主导地位。
Xu等[38]基于微观尺度研究了镁单晶中{1012}孪晶的长大过程。首次提出一个棱柱面和基面的交界面(PB交界面)(见图 1),由于PB交界面形成所需能量很低,在孪晶成核后两侧尖端会出现PB交界面,PB交界面的扩展能有效促进孪晶长大。在Xu等研究基础上,Leclercq等[39]研究了镁单晶中{1101}和{1102}两种孪晶的长大机制。在整个长大过程中,{1102}孪晶长大满足Xu等提出的PB交界面扩展理论,但{1101}孪晶长大不满足PB交界面扩展理论。在{1102}和{1101}孪晶的扩展长大中,{1102}孪晶更多和内部因素有关,{1101}孪晶则更多和外在因素有关,并通过能量角度分析得出双层孪晶面滑移扩展是最主要机制。

|
图 1 镁晶胞中PB交界面的结构图和能量图[38] (a)棱柱面和基面面对面接触的PB交界图;(b)弛豫交界面,可得交界面中心带有明显位错;(c)不同孪晶面与[1011]方向夹角θ和所需能量变化曲线 Fig. 1 Structure and energy diagrams of a prismatic/basal (PB) interface[38] (a)basal and prismatic unit cells placed face to face across the PB interface; (b)relaxed structure of the interface with the core of a misfit dislocation highlighted; (c)energy curves of twin interface as a function of its angle θ with respect to the horizontal [1011] direction |
Xu等[43]通过分子动力学方法研究了镁中一次和二次孪生模式,主要涉及一个对称倾斜晶界与基面滑移的位错交互作用而引起的变形孪晶。在该研究中,一次孪晶{1121}和二次孪晶{1122}都会生成,但这两种孪晶形核与长大需要克服的能垒不同,{1121}孪晶是最易开启的孪生模式。同时这两种孪晶迁移机理也不相同,{1121}孪晶晶界迁移是由原子位置局域调整得以实现,而{1122}孪晶晶界迁移是由于孪晶界上的锥形滑移而激发的。综上所述,国内外基于分子动力学对镁合金孪晶变形机制的研究集中于拉伸孪晶的形核和长大机理,对于压缩孪晶的变形机理研究较少,有待进一步深入研究。
3.2 基于分子动力学的滑移和晶界滑移变形机制晶体滑移本质是通过位错滑移实现的,而发生位错滑移时,滑移面上的原子是逐个移动的,并非整个原子层同时移动[27]。基于分子动力学对镁合金滑移变形机制的研究已有不少报道。
Kim等[44]研究了多晶镁晶界处1/3{1011}〈1123〉和1/3{1122}〈1123〉两种〈c+a〉锥型位错的形核机理,并对这两种〈c+a〉锥型位错的结构和性质做了对比论述。Tang等[45]通过分子动力学方法研究了镁中〈c+a〉位错形核机理和滑移行为,介绍了〈c+a〉位错的分解模式及核结构。通过重构位错核结构,定量地测定了具有不同取向的〈c+a〉位错滑移所需临界剪切应力,观察到了极高的各向异性。另外,模拟中明显观察到〈c+a〉位错滑移时无扩散攀爬现象和通过交滑移由Ⅰ型锥面向Ⅱ型锥面转变的现象。
Luque等[46]研究了镁纳米柱晶体中基面滑移变形机制,比较了Liu-EAM和Sun-EAM两种不同势函数在描述镁原子间相互作用的准确性。相对于Sun-EAM势,利用Liu-EAM势表现出强的SD效应,这是因为基面上堆垛层错能和位错开动所需最小应力的差异造成的。由此可得:镁合金材料的可塑性是由预先存在的位错或孪晶的运动引起,通常与相关缺陷的固有成核无关,而与应力集中处的非均成核和第二相原子等因素有很大关系。此外,Somekawa等[47]将分子动力学方法和纳米压痕蠕变实验有效结合起来研究了镁合金中晶界结构对晶界滑移变形行为的影响。结果表明,镁晶界周围的主要变形机理为晶界滑移,晶界滑移的开动与晶界能量密切相关,晶界能量越高晶界滑动速率越高,而将铝原子加入到镁基体中降低了晶界能量,能够抑制晶界滑移的开动。
3.3 从分子动力学角度探讨镁合金塑性变形机制的影响因素镁合金塑性变形机制主要受温度、合金元素、晶体取向、变形速率和晶粒尺寸等因素影响。基于分子动力学方法解析镁合金塑性变形机制若干影响因素的作用机理是近年来的研究热点。
合金元素对镁合金塑性变形的影响主要表现在三个方面:一是改变合金广义层错能;二是促使镁合金晶体结构发生变化;三是通过改变合金的相结构及分布情况影响镁合金塑性变形模式。Karewar等[48]研究了Li元素对纳米晶体镁塑性变形的影响。模拟过程选用Mg-Li EAM势函数[49],它是基于CD-EAM势函数[50]提出的。当加入Li元素时,降低了纳米晶体镁室温下塑性变形时的各向异性,提高了延展性和成形性,其原因是促进了〈c+a〉锥面滑移和{1012}拉伸孪晶的生长,并抑制了基面滑移和{1011}, {1013}压缩孪晶系的开动。
Somekawa等[51]研究了添加合金元素对镁合金晶界滑移的影响。结果表明,向镁中添加Al和Ag等元素可以抑制晶界滑移,Ag的添加对于抑制晶界滑移更有效;他们还研究了晶界结构对晶界的塑性变形机理和能量变化的影响,发现倾斜角为23°比倾斜角为78°的晶界具有更高的界面能,得出在晶界处更容易发生晶界滑移,这也验证了纳米压痕蠕变实验[48]的结果。
Zu等[52]基于纳观尺度研究了不同取向镁单晶拉伸和压缩变形行为。如图 2所示,载荷加载方向沿y轴始终保持垂直于[1010]轴方向,θ表示y轴和[1210]轴夹角,角度范围从0°~90°选取了10种不同取向进行研究。结果显示:当加载方向从[0001]轴起接近45°时,由于高的Schmid因子和低的CRSS值,在拉伸和压缩变形中基面位错主导初始塑性变形;当加载方向接近[1210]轴时,基面滑移被抑制,锥面滑移和孪晶变形机制开动主导塑性变形,而且沿[1210]方向拉伸和压缩的屈服强度值远高于其他取向的屈服强度值。

Liao等[53]利用分子动力学模拟了镁中析出相Mg17Al12对基面滑移的影响。势函数选用Jelinek等[54]开发的MEAM势函数,和EAM相比较,MEAM考虑了角向电子密度,因而表现出更好的精度。模拟结果如图 3所示:基面位错能够通过析出相,而不会产生包围析出相的位错环,这与著名的Orowan变形机制不同;基面位错滑移通过析出相-基体界面,基体被基面位错塑性剪切,析出相仅发生弹性剪切而没有塑性剪切。

在微纳米尺度,现有的分子动力学研究多集中于镁单晶体系,很难准确模拟和预测镁合金宏观各向异性力学性能,无法满足其实际成形过程的需要。因此,如何将分子动力学模拟应用于镁合金宏观塑性成形是目前该领域的难点问题,为此,大量研究工作有待深入和系统地进行。
(1) 势函数的精确性对计算结果精度产生直接影响,努力开发高精度、高效率和高扩展性的势函数一直是科研人员追求的目标。然而,获得尽可能准确而形式又相对简单的势函数始终无法兼顾。随着计算机技术的发展,将第一性原理计算、量子化学分析和参数拟合等研究方法有效结合,开发适用于镁合金二元甚至多元体系的势函数将成为未来该领域的研究热点。
(2) 在当前镁单晶体系塑性变形研究的基础上,模拟不同组分、不同浓度的多元镁合金体系塑性变形机制,同时加强对镁合金塑性变形机制影响因素的研究将成为未来该领域的另一个热点问题。目前,分子动力学模型原子数相对较少,为了提高其动力学和热学性质模拟精度,未来应建立更大规模的计算模型。
(3) 基于微纳米尺度的研究,可以为更高尺度模拟提供理论依据和技术准则。因此,未来应加大微纳观尺度下镁合金塑性变形机理的研究力度,同时要寻求和建立镁合金从原子排列到相的变化到材料宏观性能的相互关系。通过实现纳观、微观、介观、宏观不同尺度之间的衔接,构建多尺度模型研究材料的成分-结构-性能之间的关系,从而缩短材料的研发周期并降低研发成本。
| [1] |
宋珂. 镁合金在汽车轻量化中的应用发展[J].
机械研究与应用, 2007, 20 (1): 14–16.
SONG K. The development and application of magnesium alloys in automotive industry[J]. Mechanical Research & Application, 2007, 20 (1): 14–16. DOI: 10.3969/j.issn.1007-4414.2007.01.008 |
| [2] |
胡斌, 彭立明, 曾小勤, 等. 镁合金在汽车领域中的应用(一)——镁合金在汽车领域中应用背景和发展现状[J].
铸造工程, 2007, 31 (4): 34–39.
HU B, PENG L M, ZENG X Q, et al. Applications of magnesium alloy in automobile:background and developmental status of the magnesium alloy in automobile[J]. Foundry Engineering, 2007, 31 (4): 34–39. DOI: 10.3969/j.issn.1673-3320.2007.04.010 |
| [3] |
吴国华, 陈玉狮, 丁文江. 镁合金在航空航天领域研究应用现状与展望[J].
载人航天, 2016, 22 (3): 281–292.
WU G H, CHEN Y S, DING W J. Current research, application and future prospect of magnesium alloys in aerospace industry[J]. Manned Spaceflight, 2016, 22 (3): 281–292. DOI: 10.3969/j.issn.1674-5825.2016.03.002 |
| [4] | WU Z, CURTIN W A. The origins of high hardening and low ductility in magnesium[J]. Nature, 2015, 526 (7571): 62–67. DOI: 10.1038/nature15364 |
| [5] | WU Z, AHMAD R, YIN B, et al. Mechanistic origin and prediction of enhanced ductility in magnesium alloys[J]. Science, 2018, 359 (6374): 447–452. DOI: 10.1126/science.aap8716 |
| [6] |
宋广胜, 陈强强, 徐勇, 等. AZ31镁合金室温拉伸微观变形机制EBSD原位跟踪研究[J].
材料工程, 2016, 44 (4): 1–8.
SONG G S, CHEN Q Q, XU Y, et al. Deformation micro-mechanism of AZ31 Mg alloy during tension at room temperature by EBSD in-situ tracking[J]. Journal of Materials Engineering, 2016, 44 (4): 1–8. DOI: 10.3969/j.issn.1673-1433.2016.04.001 |
| [7] |
丁文江, 曾小勤. 中国Mg材料研发与应用[J].
金属学报, 2010, 46 (11): 1450–1457.
DING W J, ZENG X Q. Research and applications of magnesium in China[J]. Acta Metallurgica Sinica, 2010, 46 (11): 1450–1457. |
| [8] | SOLONENKO O P, KUDINOV V V, SMIRNOV A V, et al. Micro-metallurgy of splats:theory, computer simulation and experiment[J]. JSME International Journal, 2005, 48 (3): 366–388. DOI: 10.1299/jsmeb.48.366 |
| [9] | POKORSKA I. Computer methods in design and identification of powder metallurgy materials[J]. Advanced Materials Research, 2011, 314/316 : 1666–1669. DOI: 10.4028/www.scientific.net/AMR.314-316.1666 |
| [10] |
李立云, 曲周德. 镁合金塑性变形机制及动态再结晶研究进展[J].
机械研究与应用, 2015, 28 (6): 197–199.
LI L Y, QU Z D. Research achievements of plastic deformation mechanism and dynamic recrystallization of magnesium alloy[J]. Mechanical Research & Application, 2015, 28 (6): 197–199. |
| [11] |
陈振华.
变形镁合金[M]. 北京: 化学工业出版社, 2005: 48-98.
CHEN Z H. Wrought magnesium alloys[M]. Beijing: Chemical Industry Press, 2005: 48-98. |
| [12] |
刘庆. 镁合金塑性变形机制研究进展[J].
金属学报, 2010, 46 (11): 1458–1472.
LIU Q. Research progress on plastic deformation mechanism of Mg alloys[J]. Acta Metallurgica Sinica, 2010, 46 (11): 1458–1472. |
| [13] |
詹美燕, 李春明, 尚俊玲. 镁合金的塑性变形机制和孪生变形研究[J].
材料导报, 2011, 25 (2): 1–7.
ZHAN M Y, LI C M, SHANG J L. Investigation of the plastic deformation mechanism and twinning of magnesium alloys[J]. Materials Review, 2011, 25 (2): 1–7. |
| [14] | AGNEW S R, DUYGULU O. Plastic anisotropy and the role of non-basal slip in magnesium alloy AZ31B[J]. International Journal of Plasticity, 2005, 21 (6): 1161–1193. DOI: 10.1016/j.ijplas.2004.05.018 |
| [15] | KOIKE J, KOBAYASHI T, MUKAI T, et al. The activity of non-basal slip systems and dynamic recovery at room temperature in fine-grained AZ31B magnesium alloys[J]. Acta Materialia, 2003, 51 (7): 2055–2065. DOI: 10.1016/S1359-6454(03)00005-3 |
| [16] | KAISER F, LETZIG D, BOHLEN J, et al. Anisotropic properties of magnesium sheet AZ31[J]. Materials Science Forum, 2003, 419/422 : 315–320. DOI: 10.4028/www.scientific.net/MSF.419-422.315 |
| [17] | ANDO S, TANAKA M, TONDA H. Pyramidal slip in magne-sium alloy single crystals[J]. Materials Science Forum, 2003, 419/422 : 87–92. DOI: 10.4028/www.scientific.net/MSF.419-422.87 |
| [18] |
刘俊伟, 陈振华, 陈鼎, 等. 孪生对热轧AZ31镁合金中低温变形行为的影响[J].
航空材料学报, 2012, 32 (1): 10–14.
LIU J W, CHEN Z H, CHEN D, et al. Effect of twinning on moderate-temperature deformation behavior of hot-rolled Mg alloy[J]. Journal of Aeronautical Materials, 2012, 32 (1): 10–14. DOI: 10.3969/j.issn.1005-5053.2012.1.003 |
| [19] | YOO M H. Slip, twinning, and fracture in hexagonal close-packed metals[J]. Metallurgical Transactions A, 1981, 12 (3): 409–418. DOI: 10.1007/BF02648537 |
| [20] | KESHAVARZ Z, BARNETT M R. EBSD analysis of deform-ation modes in Mg-3Al-1Zn[J]. Scripta Materialia, 2006, 55 (10): 915–918. DOI: 10.1016/j.scriptamat.2006.07.036 |
| [21] | WANG Y N, HUANG J C. The role of twinning and untwining in yielding behavior in hot-extruded Mg-Al-Zn alloy[J]. Acta Materialia, 2007, 55 (3): 897–905. DOI: 10.1016/j.actamat.2006.09.010 |
| [22] | LOU X Y, LI M, BOGER R K, et al. Hardening evolution of AZ31B Mg sheet[J]. International Journal of Plasticity, 2007, 23 (1): 44–86. DOI: 10.1016/j.ijplas.2006.03.005 |
| [23] | KOIKE J. Enhanced deformation mechanisms by anisotropic plasticity in polycrystalline Mg alloys at room temperature[J]. Metallurgical and Materials Transactions A, 2005, 36 (7): 1689–1696. DOI: 10.1007/s11661-005-0032-4 |
| [24] | BARNETT M R, KESHAVARZ Z, BEER A G, et al. Non-Schmid behavior during secondary twinning in a polycrystalline magnesium alloy[J]. Acta Materialia, 2008, 56 (1): 5–15. DOI: 10.1016/j.actamat.2007.08.034 |
| [25] | BARNETT M R. Twinning and the ductility of magnesium allo-ys:part Ⅰ:"Tension" twins[J]. Materials Science and Engineering:A, 2007, 464 (1/2): 1–7. |
| [26] | BARNETT M R. Twinning and the ductility of magnesium allo-ys:part Ⅱ:"Contraction" twins[J]. Materials Science and Engineering:A, 2007, 464 (1/2): 8–16. |
| [27] | CIZEK P, BARNETT M R. Characteristics of the contraction twins formed close to the fracture surface in Mg-3Al-1Zn alloy deformed in tension[J]. Scripta Materialia, 2008, 59 (9): 959–962. DOI: 10.1016/j.scriptamat.2008.06.041 |
| [28] |
余琨, 黎文献, 王日初. 镁合金塑性变形机制[J].
中国有色金属学报, 2005, 15 (7): 1081–1086.
YU K, LI W X, WANG R C. Plastic deformation mechanism of magnesium alloys[J]. The Chinese Journal of Nonferrous Metals, 2005, 15 (7): 1081–1086. DOI: 10.3321/j.issn:1004-0609.2005.07.016 |
| [29] |
郑翊, 严红革, 陈吉华, 等. 高应变速率轧制ZK60板材的超塑性行为[J].
中国有色金属学报, 2014, 24 (4): 839–847.
ZHENG Y, YAN H G, CHEN J H, et al. Superplasticity behavior of ZK60 alloy sheet prepared by high strain rate rolling process[J]. The Chinese Journal of Nonferrous Metals, 2014, 24 (4): 839–847. |
| [30] |
殷开梁.分子动力学模拟的若干基础应用和理论[D].杭州: 浙江大学, 2006. YIN K L.Some basic applications and theories of molecular dynamics simulation[D].Hangzhou: Zhejiang University, 2006. |
| [31] | ABRAHAM F F, WALKUP R, GAO H, et al. Simulating mat-erials failure by using up to one billion atoms and the world's fastest computer:brittle fracture[J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99 (9): 5777–5782. DOI: 10.1073/pnas.062012699 |
| [32] |
段献宝.晶格反演修正型嵌入原子势函数理论及应用研究[D].武汉: 华中科技大学, 2015. DUAN X B.Theory and applications of lattice inversion modified embedded atom method[D].Wuhan: Huazhong University of Science and Technology, 2015. |
| [33] | LIU X Y, ADAMS J B, ERCOLESSI F, et al. EAM potential for magnesium from quantum mechanical forces[J]. Modelling and Simulation in Materials Science and Engineering, 1996, 4 (3): 293–303. DOI: 10.1088/0965-0393/4/3/004 |
| [34] | SUN D Y, MENDELEV M I, BECKER C A, et al. Crystal-melt interfacial free energies in hcp metals:a molecular dynamics study of Mg[J]. Physical Review B, 2006, 73 : 024116. DOI: 10.1103/PhysRevB.73.024116 |
| [35] | BASKES M I, JOHNSON R A. Modified embedded atom poten-tials for HCP metals[J]. Modelling Simulation in Materials Science Engineering, 1994, 2 (1): 147–163. DOI: 10.1088/0965-0393/2/1/011 |
| [36] | KIM K H, JEON J B, LEE B J. Modified embedded-atom meth-od interatomic potentials for Mg-X(X=Y, Sn, Ca) binary systems[J]. Calphad, 2015, 48 : 27–34. DOI: 10.1016/j.calphad.2014.10.001 |
| [37] | KIM K H, LEE B J. Modified embedded-atom method interato-mic potentials for Mg-Nd and Mg-Pb binary systems[J]. Calphad, 2017, 57 : 55–61. DOI: 10.1016/j.calphad.2017.03.003 |
| [38] | XU B, CAPOLUNGO L, RODNEY D. On the importance of prismatic/basal interfaces in the growth of {1012} twins in hexagonal close packed crystals[J]. Scripta Materialia, 2013, 68 (11): 901–904. DOI: 10.1016/j.scriptamat.2013.02.023 |
| [39] | LECLERCQ L, CAPOLUNGO L, RODNEY D. Atomic-scale comparison between {1101} and {1102} twin growth mecha-nisms in magnesium[J]. Materials Research Letters, 2014, 2 (3): 152–159. DOI: 10.1080/21663831.2014.880548 |
| [40] | WANG J, BEYERLEIN I J, TOMÉ C N. An atomic and probabilistic perspective on twin nucleation in Mg[J]. Scripta Materialia, 2010, 63 (7): 741–746. DOI: 10.1016/j.scriptamat.2010.01.047 |
| [41] | WANG J, HIRTH J P, TOMÉ C N. (1012) twinning nucleation mechanisms in hexagonal-close-packed crystals[J]. Acta Materi-alia, 2009, 57 (18): 5521–5530. DOI: 10.1016/j.actamat.2009.07.047 |
| [42] | AGHABABAEI R, JOSHI S P. Micromechanics of tensile twinning in magnesium gleaned from molecular dynamics simul-ations[J]. Acta Materialia, 2014, 69 (5): 326–342. |
| [43] | XU H L, SU X M, YUAN G Y, et al. Primary and secondary modes of deformation twinning in HCP Mg based on atomistic simulations[J]. Transactions of Nonferrous Metals Society of China, 2014, 24 (12): 3804–3809. DOI: 10.1016/S1003-6326(14)63536-6 |
| [44] | KIM D H, EBRAHIMI F, MANUEL M V, et al. Grain-boundary activated pyramidal dislocations in nano-textured Mg by mole-cular dynamics simulation[J]. Materials Science and Engin-eering:A, 2011, 528 (16): 5411–5420. |
| [45] | TANG Y Z, JAAFAR A, AWADY E. Formation and slip of pyramidal dislocations in hexagonal close-packed magnesium single crystals[J]. Acta Materialia, 2014, 71 : 319–332. DOI: 10.1016/j.actamat.2014.03.022 |
| [46] | LUQUE A, GHAZISAEIDI M, CURTIN W A. Deformation modes in magnesium (0001) and (0111) single crystals:simulations versus experiments[J]. Modelling and Simulation in Materials Science and Engineering, 2013, 21 : 045010. DOI: 10.1088/0965-0393/21/4/045010 |
| [47] | SOMEKAWA H, MUKAI T. Effect of grain boundary structures on grain boundary sliding in magnesium[J]. Materials Letters, 2012, 76 (1): 32–35. |
| [48] | KAREWAR S, GUPTA N, GROH S, et al. Effect of Li on the deformation mechanisms of nanocrystalline hexagonal close pack-ed magnesium[J]. Computational Materials Science, 2017, 126 : 252–264. DOI: 10.1016/j.commatsci.2016.09.002 |
| [49] | KAREWAR S V, GUPTA N, CARO A, et al. A concentration dependent embedded atom method potential for the Mg-Li system[J]. Computational Materials Science, 2014, 85 : 172–178. DOI: 10.1016/j.commatsci.2013.12.037 |
| [50] | CARO A, CROWSON D A, CARO M. Classical many-body potential for concentrated alloys and the inversion of order in iron-chromium alloys[J]. Physical Review Letters, 2005, 95 (7): 075702. DOI: 10.1103/PhysRevLett.95.075702 |
| [51] | SOMEKAWA H, MUKAI T. Molecular dynamics simulation of grain boundary plasticity in magnesium and solid-solution magnesium alloys[J]. Computational Materials Science, 2013, 77 (3): 424–429. |
| [52] | ZU Q, GUO Y F, XU S, et al. Molecular dynamics simulations of the orientation effect on the initial plastic deformation of magnesium single crystals[J]. Acta Metallurgica Sinica, 2016, 29 (3): 301–312. |
| [53] | LIAO M, LI B, HORSTEMEYER M F. Interaction between bas-al slip and a Mg17Al12 precipitate in magnesium[J]. Metallurgical and Materials Transactions A, 2014, 45 (8): 534–539. |
| [54] | JELINEK B, GROH S, HORSTEMEYER M F, et al. Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys[J]. Physical Review B, 2012, 85 : 245102. DOI: 10.1103/PhysRevB.85.245102 |