 2019, Vol. 47
2019, Vol. 47文章信息
- 卢子龙, 安立宝, 刘扬
- LU Zi-long, AN Li-bao, LIU Yang
- 不同浓度硼掺杂石墨烯吸附多层金原子的第一性原理研究
- First principles study of adsorption of multilayer gold atoms on graphene doped with B under various concentrations
- 材料工程, 2019, 47(4): 64-70
- Journal of Materials Engineering, 2019, 47(4): 64-70.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2018.000208
-
文章历史
- 收稿日期: 2018-03-01
- 修订日期: 2019-03-15




石墨烯作为新兴碳纳米材料,具有大的比表面积、良好的化学稳定性、优异的电学和力学性能等诸多优点,因此其在柔性电极、电化学传感器和新能源电池材料等领域有着广阔的应用前景[1-2]。然而,石墨烯与金属间的接触电阻通常较高,导致额外的发热和能耗,降低器件的性能和使用寿命[3]。
降低石墨烯接触电阻的方法很多,传统的方法包括高温退火法、局部焦耳热法、超声波焊接法等[4-5],但这些方法存在可重复性差、技术不够完善、对器件产生损伤等缺点,因此需进一步改进。研究表明,掺杂可有效地提高碳纳米材料的电子输运性能,降低接触电阻。本课题组通过介电泳方法将Au掺杂碳纳米管(CNT)组装到两金属电极之间进行测量,发现Au掺杂后CNT的接触电阻较本征CNT降低约50%[4]。半导体材料与金属电极接触时,由于两者费米能级的不同导致接触界面存在肖特基势垒,掺杂可以调节半导体材料的电子能带结构,降低界面势垒高度[6]。同时,掺杂剂作为电子的供体和受体,可提高掺杂处的局部电子浓度,这不仅可以提高半导体材料自身的导电性,还可以增强金属对半导体的浸润性[7],提高金属与半导体的接触面积,进而增加导电通道,降低半导体材料与金属间的接触电阻[8]。
对于石墨烯而言,B元素是良好的掺杂剂,它可以将空穴引入石墨烯中,形成p型掺杂[9],进而改变石墨烯的电子能带结构,重新分配石墨烯的电子分布[10]。同时,B作为电子的受体可以改良石墨烯表面的吸附性能和电子传递速率[11]。Au元素化学性质稳定,是研究中常用的电极材料。Gahoi等[12]在95%氩气和5%氢气环境下对Au, Ni, Au-Ni合金, Pd, Pt-Au合金等5种金属或合金与单层石墨烯进行高温处理1h形成复合材料,发现Au与石墨烯的电阻率最小,为92Ω·μm。
利用密度泛函理论(DFT)研究掺杂石墨烯的电接触特性,可以从微观层面对其接触电阻的形成和降阻原理作出解释,同时减少实验成本,缩短研究周期。在以往的研究中,吸附模型多为石墨烯吸附单个或有限数量的单层金属原子,实际上在应用中石墨烯都是与多层金属原子发生交互作用。因此,本工作利用第一性原理模拟研究不同浓度B掺杂石墨烯与多层Au原子间的吸附作用和界面电学性能。首先计算不同浓度B掺杂石墨烯的结构稳定性,再将多层Au原子置入掺杂石墨烯体系中,计算B掺杂石墨烯吸附多层Au原子的吸附能、态密度、电荷密度和电荷转移量,研究B掺杂浓度对石墨烯吸附多层Au原子的影响。
1 物理模型与计算方法本工作选用超晶胞为6×6×1的单层石墨烯,共72个C原子。掺杂方式为晶格掺杂,即在石墨烯模型中将C原子替换为B原子,采用每两个掺杂B原子连接一个C原子的掺杂结构。研究表明该结构的结合能较大,体系较稳定[13]。为了后期模拟吸附时,使多层Au原子与B原子形成更多有效的接触,掺杂位置尽量分布于石墨烯的中心区域,掺杂模型如图 1 (a)所示。将超晶胞为2×1×1的多层Au原子(3层共23个Au原子)置入石墨烯上方构造吸附模型,如图 1 (b)所示。为了避免不同晶胞间的相互作用影响计算结果,建立1.278nm×1.223nm×1.089nm的真空层作为周期结构的边界条件。掺杂的浓度分别为1.39% (1个B原子), 4.17% (3个B原子), 6.94% (5个B原子), 9.72% (7个B原子), 12.50% (9个B原子)和15.28% (11个B原子)。除了本征石墨烯用G表示之外,按照上述浓度掺杂将各掺杂石墨烯命名为G-1B,G-3B,G-5B,G-7B,G-9B和G-11B。本工作首先对石墨烯掺杂模型进行结构优化,在此基础上置入多层Au原子形成吸附模型,并对吸附模型进行结构优化和能量计算,最后分析不同浓度B掺杂对石墨烯吸附多层Au原子结构稳定性和电学性能的影响。图 2为优化后的掺杂吸附模型,可以看出,结构优化后吸附模型中掺杂石墨烯的几何形变,随着掺杂浓度的提高而增大,但掺杂浓度不大于12.50%时,石墨烯弯曲角度均小于30°;当掺杂浓度为15.28%时,掺杂石墨烯表面褶皱明显,弯曲角度达50°,产生几何畸变,且通过后续计算表明,该掺杂浓度下吸附模型的赝能隙较低,体系的共价键强度较低,所以不再讨论。

|
图 1 石墨烯掺杂模型(a)和吸附模型(b) Fig. 1 Doping model(a) and adsorption model(b) of graphene |

|
图 2 掺杂石墨烯吸附多层Au原子模型 (a)G-3B-Au;(b)G-7B-Au;(c)G-11B-Au Fig. 2 Model of doped graphene adsorbing multilayer Au atoms (a)G-3B-Au; (b)G-7B-Au; (c)G-11B-Au |
仿真计算以第一性原理为基础,通过广义梯度近似(GGA)进行结构优化和能量计算,其中交换相关函数选择PBE函数[14-15];原子轨道计算采用双数值轨道基矢(DNP)。优化的过程是一个自洽迭代的计算过程,当输出的电子密度函数与其前一次的计算结果差值小于一定收敛精度时,认为得到自洽结果[16],截止半径为0.03nm。其他收敛标准如下:单原子能量收敛标准为1.0×10-5 Ha (1Ha=27.212eV),原子间最大相互作用力收敛标准为0.02Ha/nm,原子间最大位移收敛标准为0.0005nm。当计算结果达到以上标准时优化结束。
2 结果与讨论 2.1 掺杂模型的结构稳定性结合能是指原子由自由状态形成化合物所释放的能量。通过计算体系的结合能可以判断掺杂后石墨烯结构的稳定性,其计算公式为[17]:
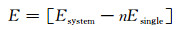
|
(1) |
式中:E为体系结合能; Esystem为体系总能量; n为体系中所包含原子的总数; Esingle为体系中所有原子在孤立状态下的原子总能。结合能为负值说明原子从孤立状态形成结合物时放出能量,放出的能量越多,从热力学角度说明物质的几何结构越稳定[17]。
不同浓度B掺杂石墨烯结构优化后的结合能及优化后掺杂石墨烯与本征石墨烯结合能的相对变化量(ΔE)如表 1所示。B掺杂会引入空穴到石墨烯中,形成P型掺杂[10],使石墨烯的结构发生改变。正如表 1中所示,新体系的结合能较本征石墨烯均有所降低,活跃度有所提升。其中G-9B变化最大,相对减少了5.01%;G-1B变化最小,相对减少了0.51%。单层石墨烯为六边形蜂窝状结构,自身具有极好的稳定性,虽然掺杂后结合能有所减小,但减小的幅度均小于5.1%,且最低的结合能仍在450eV以上,说明B掺杂石墨烯结构可以稳定地存在。这是因为B元素与C同周期,原子半径相近,低浓度掺杂后对石墨烯原结构破坏较小。
| Doping concentration/% | E/eV | ΔE/% |
| 0 | -475.237 | - |
| 1.39 | -472.812 | -0.51 |
| 4.17 | -468.926 | -1.33 |
| 6.94 | -463.761 | -2.41 |
| 9.72 | -457.517 | -3.73 |
| 12.50 | -451.452 | -5.01 |
分析吸附能可了解多层Au原子与不同浓度B掺杂石墨烯间的相互作用情况,见表 2。吸附能的计算公式为:
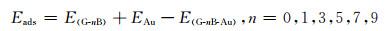
|
(2) |
| Doping concentration/% | Eads/eV |
| 0 | 0.653 |
| 1.39 | 2.395 |
| 4.17 | 3.402 |
| 6.94 | 3.538 |
| 9.72 | 5.646 |
| 12.50 | 7.592 |
式中:E(G-nB)为吸附前本征/掺杂石墨烯的总能;EAu为吸附前Au原子的总能;EG-nB-Au为吸附Au原子后的吸附体系总能。由表 2可知本征石墨烯与多层Au原子的吸附能为0.653eV,两者间交互作用很小。掺杂石墨烯中B作为电子受体,使石墨烯与富含电子的Au更容易发生交互作用[18];掺杂体系的吸附能较本征石墨烯吸附多层Au原子时平均提高了5倍以上,表明B掺杂石墨烯与多层Au原子间形成了稳定的化学吸附。
2.3 态密度分析态密度(DOS)表示电子在某一能量范围的分布情况,是能带结构的一个可视化体现。态密度图中横坐标零点处代表吸附模型的费米能级(Ef),在Ef两侧相邻的两个DOS波峰横坐标的间距为赝能隙。赝能隙直接反映了体系成键的共价性强弱,赝能隙越大,说明共价性越强[19]。图 3为各掺杂吸附模型与本征吸附模型赝能隙的对比图。从图 3可以看出,B掺杂后吸附模型的赝能隙明显增大,稳定性得到增强。赝能隙具体的数值如图 4所示,其中G-3B-Au的赝能隙最大,为1.47eV,较G-Au的赝能隙提高了约1.8倍;当掺杂浓度再提高时,赝能隙的值随之减小,G-9B-Au的赝能隙仅为0.83eV,与G-Au的赝能隙相差很小,说明掺杂浓度过高反而会降低体系成键的共价性强度。

|
图 3 本征/掺杂石墨烯吸附Au的态密度图 (a)G-Au与G-1B-Au;(b)G-Au与G-3B-Au;(c)G-Au与G-5B-Au;(d)G-Au与G-7B-Au;(e)G-Au与G-9B-Au Fig. 3 DOS of intrinsic/B-doped graphene adsorbing multilayer Au atoms (a)G-Au and G-1B-Au; (b) G-Au and G-3B-Au; (c)G-Au and G-5B-Au; (d)G-Au and G-7B-Au; (e)G-Au and G-9B-Au |

|
图 4 本征/掺杂石墨烯吸附Au的赝能隙 Fig. 4 Pseudogap of intrinsic/B-doped graphene adsorbing multilayer Au atoms |
为了进一步研究本征/掺杂石墨烯与吸附Au原子层间的成键情况,计算了本征/掺杂石墨烯中C/B原子与Au原子间的局部态密度(LDOS)。两个原子LDOS波峰在Ef附近的重合情况反映了两者间成键的强弱。经比较发现C/B原子2p轨道和Au原子6s轨道的LDOS的波峰有重叠,如图 5所示。其中B原子的2p轨道与Au原子的6s轨道的LDOS波峰重合度明显,说明B-Au的杂化效果优于C-Au,进而说明B掺杂提高了石墨烯吸附多层Au原子的能力。其中G-5B-Au, G-7B-Au和G-9B-Au中B, Au杂化较其他模型更为显著。

|
图 5 C/B与Au原子的局部态密度图 (a)G-Au;(b)G-1B-Au;(c)G-3B-Au;(d)G-5B-Au;(e)G-7B-Au;(f)G-9B-Au Fig. 5 LDOS of C/B and Au atoms (a)G-Au; (b)G-1B-Au; (c)G-3B-Au; (d)G-5B-Au; (e)G-7B-Au; (f)G-9B-Au |
本征/掺杂石墨烯吸附多层Au原子体系的电荷密度如图 6所示,图中颜色由蓝到红表示电荷密度由低到高。当原子间的电荷密度达到100e/nm3以上表示形成了化学吸附,原子间具有较强的交互作用[20]。G-Au中Au原子与C原子之间没有明显的电荷密度重合,吸附作用较弱;G-1B-Au中Au原子与B原子仅有微弱的电荷密度重合,表明较低浓度的B掺杂对电荷密度的影响较小;随着掺入B原子数量的增多,G-3B-Au, G-5B-Au, G-7B-Au和G-9B-Au中Au原子与B原子间出现明显的电荷密度重合,平均在245~272e/nm3之间,说明形成了较强的化学吸附。同时,Au原子与B原子间大量的电荷分布,有利于石墨烯与多层Au原子间的电荷转移,对降低接触电阻有显著作用。但通过比较图 6可以发现,G-9B-Au中石墨烯的几何结构较其他浓度的已有较大的变形。

|
图 6 本征/掺杂石墨烯与Au的电荷密度图 (a)G-Au;(b)G-1B-Au;(c)G-3B-Au;(d)G-5B-Au;(e)G-7B-Au;(f)G-9B-Au Fig. 6 Charge density maps of intrinsic/doped graphene and Au (a)G-Au; (b)G-1B-Au; (c)G-3B-Au; (d)G-5B-Au; (e)G-7B-Au; (f)G-9B-Au |
计算石墨烯与Au原子层之间的电荷转移量可以判断两者间电子得失情况,直接反映掺杂对G-nB-Au体系电学性能的影响。B掺杂为p型掺杂,使石墨烯中空穴数量增多,载流子浓度上升。吸附Au原子后,在B掺杂的作用下,促使电子从Au原子流向石墨烯。底层Au原子(距离石墨烯最近的一层Au原子,共8个)与B, C原子间距最小,交互作用最强。表 3为不同吸附体系中石墨烯与Au原子之间的电荷转移量。由表 3可知掺入B原子的数目越多,底层Au原子与石墨烯之间的电荷转移量越大,其中G-3B-Au,G-5B-Au,G-7B-Au和G-9B-Au的电荷转移量分别为G-Au的154.64%,163.71%,211.09%和309.07%。因此,可以看出B原子掺杂浓度对底层Au原子的电荷转移量有着显著的影响。研究石墨烯与多层Au原子整体(3层共23个Au原子)的电荷转移量时,发现G-1B-Au中的电荷转移量较本征石墨烯略有下降,这可能是因为掺入B的浓度过低,仅对底层Au原子产生了作用,而对多层Au原子整体电荷转移影响较弱。其他浓度下掺杂体系的电荷转移量随掺杂B浓度的提高而增加,说明较高浓度掺杂不仅增加底层Au原子的电荷转移量,而且可以改善多层Au原子整体的电荷转移情况,对整个体系的电学性能有所改善。
| Adsorption system | Multilayer Au atom/e | Bottom Au atom/e |
| G-Au | 1.528 | 0.496 |
| G-1B-Au | 1.424 | 0.525 |
| G-3B-Au | 1.817 | 0.767 |
| G-5B-Au | 1.894 | 0.812 |
| G-7B-Au | 1.937 | 1.047 |
| G-9B-Au | 2.198 | 1.533 |
(1) 低浓度B掺杂对石墨烯结构的稳定性影响较小,可使石墨烯吸附多层Au原子体系的吸附能平均提高5倍以上,赝能隙变宽。B原子的2p轨道和Au原子的6s轨道在费米能级处杂化作用明显,显著提高了吸附的稳定性。
(2) 在电学性能方面,随着B掺杂浓度的提高,B-Au之间的电荷密度增大,促进了石墨烯与多层Au原子间的电荷转移。当B掺杂浓度为15.28%时,石墨烯的几何结构变形过大,无实际研究价值。B掺杂浓度为6.94%和9.72%时,掺杂结合能分别为-463.761eV和-457.517eV,掺杂石墨烯的结构稳定;吸附模型的吸附能分别为3.538eV和5.646eV,且B, Au原子间杂化效果明显,表明掺杂石墨烯与Au原子形成了较稳定的化学吸附;此时,石墨烯与底层Au原子间的电荷转移量分别为本征时的163.71%和211.09%。
(3) B掺杂浓度为6.94%和9.72%时,降低石墨烯与多层Au原子间接触电阻的效果较为明显,对降低石墨烯与Au电极间的接触电阻有一定的理论指导。
| [1] | AKINWANDE D, PETRONE N, HONE J. Two-dimensional fle-xible nanoelectronics[J]. Nature Communications, 2014, 5 : 5678-1–5678-12. |
| [2] | NOVOSELOV K S, FAL'KO V I, COLOMBO L, et al. A roadmap for graphene[J]. Nature, 2012, 490 (7419): 192–200. DOI: 10.1038/nature11458 |
| [3] | NEMEC N, TOMNEK D, CUMIBERTI G. Contact dependence of carrier injection in carbon nanotubes:an ab initio study[J]. Physi-cal Review Letters, 2006, 96 (7): 076802–1. DOI: 10.1103/PhysRevLett.96.076802 |
| [4] |
刁加加, 常春蕊, 张志明, 等. 金掺杂降低碳纳米管接触电阻的实验研究[J].
材料研究学报, 2017, 31 (7): 511–516.
DIAO J J, CHANG C R, ZHANG Z M, et al. Reducing contact resistance of carbon nanotubes by Au doping[J]. Chinese Journal Of Materials Research, 2017, 31 (7): 511–516. |
| [5] | ASAKA K, KARITA M, SAITO Y. Modification of interface structure and contact resistance between a carbon nanotube and a gold electrode by local melting[J]. Applied Surface Science, 2011, 257 (7): 2850–2853. DOI: 10.1016/j.apsusc.2010.10.079 |
| [6] | KOM K K, BAE J J, PARK H K, et al. Fermi level engineering of single-walled carbon nanotubes by AuCl3 doping[J]. Journal of the American Chemical Society, 2008, 130 (38): 12757–12761. DOI: 10.1021/ja8038689 |
| [7] | SONG X.Study on mechanism of carbon nanotube/metal inter-facial bonding and related technology[D]. Shanghai: Shanghai Jiao Tong University, 2010. |
| [8] | KONG B S, JUNG D H, OH S K, et al. Single-walled carbon nanotube gold nanohybrids:application in highly effective transp-arent and conductive films[J]. The Journal of Physical Chemistry C, 2007, 111 (23): 8377–8382. DOI: 10.1021/jp071297r |
| [9] | YEH M H, LI Y S, CHEN G L, et al. Facile synthesis of boron-doped graphene nanosheets with hierarchical microstructure at atmosphere pressure for metal-free electrochemical detection of hydrogen peroxide[J]. Electrochimica Acta, 2015, 172 : 52–60. DOI: 10.1016/j.electacta.2015.01.210 |
| [10] | PULLAMSETTY A, SUBBIAH M, SUNDARA R. Platinum on boron doped graphene as cathode electrocatalyst for proton exchange membrane fuel cells[J]. International Journal of Hy-drogen Energy, 2015, 40 (32): 10251–10261. DOI: 10.1016/j.ijhydene.2015.06.020 |
| [11] | CHEN X, YANG G, FENG S, et al. Au@Au Pt nanoparticles embedded in B-doped graphene:a superior electrocatalyst for determination of rutin[J]. Applied Surface Science, 2017, 402 : 232–244. DOI: 10.1016/j.apsusc.2017.01.087 |
| [12] | GAHOI A, WAGNER S, BABLICH A, et al. Contact resistance study of various metal electrodes with CVD graphene[J]. Solid-State Electronics, 2016, 125 : 234–239. DOI: 10.1016/j.sse.2016.07.008 |
| [13] | LIU Z, ZHANG Y, WANG B, et al. DFT study on Al-doped defective graphene towards adsorption of elemental mercury[J]. Applied Surface Science, 2017, 427 : 547–553. |
| [14] | PERDEW J P, CHEVARY J A, VOSKO S H, et al. Atoms, molecules, solids, and surfaces:applications of the generalized gradient approximation for exchange and correlation[J]. Physical Review B:Condensed Matter, 1992, 46 (11): 6671–6687. DOI: 10.1103/PhysRevB.46.6671 |
| [15] | PERDEW J P, BURKE K, EMZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77 (18): 3865–3868. DOI: 10.1103/PhysRevLett.77.3865 |
| [16] |
代利峰, 刘涛, 龚亮, 等. Fe掺杂碳纳米管吸附Al原子的第一性原理研究[J].
中国有色金属学报, 2017, 27 (6): 1182–1188.
DAI L F, LIU T, GONG L, et al. First principles study of adsorption of Al atoms on Fe doped carbon nanotubes[J]. The Chinese Journal of Nonferrous Metals, 2017, 27 (6): 1182–1188. |
| [17] | PENG M M, LAI W S. Interaction between vacancies and the α-Fe/Y2O3 interface:a first-principles study[J]. Nuclear Instru-ments & Methods in Physics Research, 2015, 352 : 67–71. |
| [18] | ZHANG Y, SUN R, LUO B, et al. Boron-doped graphene as high-performance electrocatalyst for the simultaneously electro-chemical determination of hydroquinone and catechol[J]. Electro-chimica Acta, 2015, 156 : 228–234. DOI: 10.1016/j.electacta.2014.12.156 |
| [19] |
王欣, 王发展, 雷哲锋, 等. N-M (Cd, Mg)共掺闭口氧化锌纳米管场发射第一性原理研究[J].
物理学报, 2013, 62 (12): 123101-1–123101-6.
WANG X, WANG F Z, LEI Z F, et al. First-principles study of field emission properties for ZnO nanotuber capped and codoped[J]. Acta Physica Sinica, 2013, 62 (12): 123101-1–123101-6. |
| [20] | WANG K P, SHI C S, ZHAO N Q, et al. First-principle study of the effect of boron (nitrogen)-doping on adsorbing character-istics of aluminum on single-walled carbon nanotubes[J]. Acta Physica Sinica, 2008, 57 (12): 7833–7840. |