
文章信息
- 张瑾, 苏克和, 马咏梅, 曾庆丰, 成来飞, 张立同. 2015.
- ZHANG Jin, SU Ke-he, MA Yong-mei, ZENG Qing-feng, CHENG Lai-fei, ZHANG Li-tong. 2015.
- 先驱体制备典型陶瓷(C,SiC和BxC)的化学反应机理研究
- Chemical Reaction Mechanism of Typical Ceramics (C, SiC and BxC) Produced from Their Precursors
- 材料工程, 43(10): 102-112
- Journal of Materials Engineering, 43(10): 102-112.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2015.10.016
-
文章历史
- 收稿日期:2014-02-25
- 修订日期:2014-12-10




2. 西北工业大学 超高温结构复合材料国家级重点实验室, 西安 710072
2. Science and Technology on Thermostructural Composite Materials Laboratory, Northwestern Polytechnical University, Xi'an 710072, China
纤维增韧碳化硅(SiC)陶瓷基复合材料具有耐高温、低密度、高比强、韧性好和抗氧化烧蚀等优异性能,同时具有类似金属的断裂行为,对裂纹不敏感和不发生灾难性损毁等特点,被普遍认为是最具有潜力的高温热结构材料之一,在航空、航天、能源等尖端科技领域,都有着重要的战略意义[1, 2, 3]。该材料乃至构件的一体化、产业制造技术“在我国已获得全面突破”“应用研究取得重大进展”。化学气相沉积/渗透(CVD/CVI)是制备SiC基复合材料的一种最常用的方法[4]。其基本过程是在纤维编织体上相继进行碳过渡层和碳化硅基体。其中C、SiC乃至其他功能性组元(如自愈合相BxC)等,来源于先驱体烃类(甲烷、丙烯或芳香烃)、三氯甲基硅烷(CH3SiCl3,简称MTS或TMS等)和BCl3+CH4(或BCl3+丙烯)等在载气(一般为H2)和稀释气体(如Ar)中的高温热分解[5, 6, 7, 8, 9, 10]。
用CVD/CVI方法制备陶瓷基复合材料的反应体系中,其化学反应十分复杂。揭示这些组元材料的化学反应机理,对于其成分控制和工艺参数优化都有着重要的科学意义和工程价值。但由于反应体系往往处于高温环境,体系中又存在众多高活性中间体和自由基,其浓度很低,寿命很短,一般难以在线检测。因此,目前还很难通过实验方法原位研究这些反应体系的化学反应机理。
事实上,CVD/CVI的化学反应会涉及大量的中间体、过渡态、激发态甚至离子态物质,采用以量子力学为代表的理论手段可以精确地研究这些物质的结构与能量。量子力学方法也被称为第一性原理(First principles)方法。该方法只需一些基本物理量,即Plank常数h、电子质量me、电子电量e以及真空光速c、真空介电常数ε和圆周率π等,即可在几个近似(即忽略体系内各种相互作用与时间的关系,忽略原子核运动和忽略电子的相对论效应)下,求解如薛定谔(Schrdinger)方程,获得分子的电子结构、波函数、分子结构和能量等。如果分子量不是很大,组成分子的元素的原子序数也不大,结合统计热力学方法,还能够以“不含糊”(Unambiguous)的定量精度获得分子的各种热力学数据[11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31]。
研究反应机理,也就是研究反应的化学热力学和化学动力学。热力学提供反应发生的必要条件,同时也可揭示反应物、各种中间体和产物在一定条件下的平衡浓度分布。对于所需要的产物(包括凝聚态物质),还可确定其必要的生成条件,如温度、压力和进气比等。反应动力学的研究,首先可以确定反应历程,即对于先驱体是如何逐渐变成产物的化学反应过程,给出科学的认识;其次,可以确定基元反应速率常数,进而获得整体的反应速率,揭示反应物随时间的消耗量,或者产物随时间的生成量的科学机制。反应动力学的研究,可以进一步为工艺优化和质量控制提供合理的理论模型,为进一步的研究或工程应用提供理论指导。
本文针对先驱体制造典型陶瓷(C、SiC和BxC)的几个反应体系,分别叙述用量子化学等方法研究它们的反应热力学和反应动力学的理论方法和进展。这些反应体系是:用丙烯作为先驱体制备热解碳PyC,用MTS热解制备碳化硅基体,用BCl3+CH4及BCl3+丙烯制备BxC。
1 反应热力学开展反应热力学研究的前提是具备完整、可靠的热力学数据。但目前,存在大量热力学数据无从查找,或可靠性不高(不确定值大),甚至不少数据本身是错误的,而错误还不为人所知等问题[11, 12, 13]。因此,开展反应热力学研究的关键是获得反应体系中所有可能物质的可靠热力学数据。高级别的量子化学从头算方法,目前一般都可获得可靠的电子能量。尤其是基于高级别从头算的著名的G1-G4[11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21]、CBS[22, 23, 24, 25, 26, 27, 28, 29, 30, 31]等理论方法,在一般的小分子参加的反应能量计算中,已经达到可以与精确的实验测定相当的精度,而被广泛用于预测评价热力学数据。
最早,Ge等[32]研究了MTS-H2热解制备SiC基体CVD气相反应体系的热力学,采用MP二级微扰理论(MP2)确定了MTS-H2体系热解反应中的50个气态物种的结构,用高级别(耦合簇理论CCSD(T)/aug-cc-pVTZ)量子化学方法,得到了各物种的电子能量,用统计热力学方法获得了0~2000 K的热化学数据,归纳出50个物质所组成的114个化学反应,并获得了不同温度下反应的ΔrH、ΔrS和ΔrG。该工作,虽是借助于理论方法研究制备SiC的CVD反应热力学的最早工作,但在方法上还存在一定的缺陷。例如,计算方法虽属高级别理论方法,但还缺乏严格的检验;统计热力学处理中振动配分函数的谐性近似是否合理,忽略电子激发配分函数是否可靠等。为此,Deng等[33]以SiC(g)为例,用几种经过严格检验的理论方法和实验数据的对比等,确定了振动配分函数的谐性近似在0~2000K范围内带来的误差可忽略,而在15000cm-1(1.860eV)以内的电子激发配分函数则需要考虑。
在获得热化学数据的基础上,对于众多物质共存的复杂化学平衡体系,按照文献[32]的方法,很难穷尽所有化学平衡,并且也不容易确定各物质的平衡浓度随温度的分布规律。但化学平衡的热力学条件中,体系总化学势(Gibbs函数)极小原理,则为这一问题的解决提供了理论依据,即:如果视凝聚态物质为纯物质,气体都为理想气体,则化学平衡条件为

式中:s是体系总的固相物种数;N是体系总物种数;p是体系总压;ni是气相第i个物种的物质的量;nicond是固相第i个物种的物质的量,它们满足的边界条件为
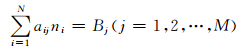
式中:aij是第i个物种中化学元素j的原子数;Bj是元素j的总原子数;M代表不同元素的总数。
基于文献[33]的结论,Yao等[34, 35]用密度泛函B3PW91/6-31G(d)确定了丙烯热解制备PyC体系的所有中间物质,用G3(MP2)和G3//B3LYP等方法获得了分子的电子能量,结合统计热力学方法获得了0~2000 K的热容和熵,用经典热力学得到了100~2000 K的ΔfH和ΔfG等。对于已有实验数据的分子,比较表明,理论计算值与实验值符合很好[36, 37]。根据式(1),结合一些芳香烃的已有实验数据,获得了如图 1所示的反应体系中各物质的浓度分布规律。由此可知,在反应体系中,固体碳(以石墨计)在约700~800 K时即可大量生成;而气相中,H2的浓度随平衡温度的升高而升高,CH4的浓度单调降低,三种稳定化合物C2H2、C2H4和C2H6具有一定的浓度分布,芳香烃的浓度都很低。图 1还给出一个值得深入研究的信息,即反应活性高的H和CH3自由基,也有较高的浓度分布,它对于反应历程中化学键的断裂——基团或原子的解离,会起重要作用。事实上,这一特性也已经在后续的动力学,即反应通道的研究中受到重视。

对于MTS-H2热解制备SiC基体CVD气相反应体系,文献[32]包括了50个中间物质。而Deng等[33, 38, 39]发现,该体系实际上至少可以包括221种气态中间物质。文献[33]首先用几种定量的精确模型化学方法[G3(MP2),CBS-Q,G3//B3LYP,G3(QCI)和QCISD(T)/aug-cc-pv5z]结合统计热力学,研究了气态SiC(X3Π)和第一激发态SiC(a1Σ)的热力学特性。结果表明,SiC从X3Π态至a1Σ的激发能和实验符合很好,SiC(X3Π)的热容(Cp,mΘ)和熵(SmΘ)与大部分文献报道结果也符合很好,但是与著名的Janaf[37]中的数据差别却很大;即使是反应物MTS,文献[40]也发现其高温热化学数据有疑问。由此说明,JANAF数据表中的个别数据并不完全可靠。文献[38]因此用G3(MP2)和G3//B3LYP结合统计热力学,对MTS-H2的气相反应体系221种气态物种的热力学数据进行了全面研究与评估,其中177个物质尚无实验数据。与已有实验数据比较,大多数理论预测结果都符合很好,但也发现个别数据有误,如C2(1Σg),C2H(2Σ),SiC(3Π),SiCl3(2A1)和CH3SiCl3(1A1)[38]。利用这些进一步完善和经过评估的数据,文献[39]研究了MTS-H2反应体系中凝聚相Si、C(石墨)和β-SiC在不同反应条件下的浓度分布,部分结果如图 2所示。这些条件是:温度300~2000K,压力6~100kPa和进气比10-2~107。图 2(a)说明,在不悬殊的进气比下,压力对凝聚态物质的生成量影响不大,而图 2(b)~(d)说明温度和进气比显著影响凝聚相的生成量。如图 2(b)所示,在1000~1500K,1~105的帽形区域内,凝聚态β-SiC的生成量才可能较大。图 2(c)说明,在600~900K和温度大于1600K都有较大量的凝聚态C生成。因此,为了避免C与SiC发生共沉积,温度区间应在1000~1500K。图 2(d)说明,凝聚态Si在进气比大于104后才逐渐生成。所以,通过调整温度和进气比,可以实现高纯凝聚态β-SiC的生成,也可实现凝聚态β-SiC与C或者与Si共沉积的技术调控。这一工作,揭示了材料制造的热力学规律,在较大和连续的实验条件范围内,为材料制造提供了理论依据。

硼碳化合物在材料的高温服役条件下,可以原位生成液态氧化硼,从而填充陶瓷基体中的孔隙和微裂纹,阻止内部材料,尤其是增韧碳纤维的氧化失效。Zeng[41]等也用经过可靠性验证的G3(MP2)和G3//B3LYP方法研究了以BCl3-CH4-H2为先驱体,用CVD法制备碳化硼的气相反应热力学,即获得了154个有关物种的热化学数据,用化学平衡原理(式(1))得到了300~2000K的所有物种的浓度分布规律。结果表明,温度高于760K时,固体B4C即可生成;当温度高于820 K时,固体C也发生共沉积,研究者同时发现BHCl3和BCH3Cl2是CVD制备碳化硼的高浓度中间体,提出反应物的初级反应不仅有BCl3+H2=BHCl2+HCl,而且还有BCl3+CH4=BCH3Cl2+HCl的新见解。Wang等[42]进一步研究了BCl3-C3H6-H2体系制备B4C气相化学反应热力学,即用DFT、G3(MP2)和统计热力学方法确定了体系中可能存在的325个物种(其中134个为新物种)的热化学数据,同样用Gibbs能量最小化原理(式(1)),获得了各物种的平衡浓度分布随温度变化的关系,表明固相石墨在反应的开始阶段(温度约400 K)即能大量生成,当温度高于700K时,B4C随温度升高而大量沉积。其中石墨的产量可通过改变进气量或总压强来调控。对于在较大范围内连续变化的反应条件,邓娟利[43]还进一步研究了这两个反应体系中凝聚态物质的生成规律,获得了一系列双变量曲面图,集中反映了各技术参数对不同材料组元生成量的影响,为特定材料组成的工艺调控提供了科学依据。
近年来,用类似的热力学理论方法,Deng等[44, 45, 46]还研究了ZrCl4-BCl3-H2体系制备过渡金属化合物ZrB[44]2、ZrCl4-BCl3-CH3SiCl3-H2-Ar体系制备ZrB和SiC的共沉积[45],以及SiCl4-NH3-H2体系制备Si3N4的气相反应的热力学[46]。Xue等[47]研究了SiCl4-NH3-CH4-H2体系制备SiC和Si3N4的共沉积热力学。Liu等[48, 49]也研究了MTS-ZrCl4-CH4-H2体系中ZrC和SiC共沉积[48]和SiCl4-NH3-C3H6-H2体系中Si-C-N共沉积[49]的气相反应热力学。这些工作,都从热力学理论上进一步揭示了材料制造的科学原理,为不同基体、不同共沉积组元、不同功能组元等的生成与抑制的技术调控,提供了理论指导。
2 反应动力学 2.1 反应通道众所周知,一个化学反应往往必须经历一系列的中间步骤(基元反应)方可完成。决定一个基元反应优先进行的关键物理量是活化能。为了揭示反应进行的具体历程,实验上需要设计十分复杂的研究方案,完成大量的中间物质的定性和定量测定,难度非常大,甚至难以完成。而理论研究则不受实验装置和条件的限制。理论方法首先系统性地确定所有可能的反应途径,从而根据能垒(活化能)最小的原则,获得最佳反应通道。
在文献[34, 35]热力学研究的基础上,Qu等[50]用密度泛函B3PW91/6-311G(d,p)、多参考态组态相互作用(MRCISD)、G3(MP2)、统计热力学和经典热力学等理论方法,研究了丙烯热解制备PyC气相反应途径。例如,丙烯具有21种不同的初级分解步骤,如图 3所示(用ΔrG数据描图)。初级反应中最有利的一步是从单重态(基态)到三重态(a3A)的激发(虚线所示),吉布斯自由能为270.2kJ·mol-1。C3H6(a3A)可进一步按5条均裂反应通道分解,所需能量在100.7~188.7kJ·mol-1之间。而其他方式,能量或能垒都在335.9kJ·mol-1以上。因此确定,在较低温度下,最先发生的反应,应该是丙烯的激发和分解,同时产生H·和CH3·等自由基。

为了准确了解反应发生的通道,对于初始分解所产生的10种不同的中间产物,文献[50]都进行了深入研究。根据所需能量或能垒最小原则,即可确定最佳反应途径。结果表明,最有利的反应,大都是按自由基参加的链反应方式进行。由此确定出C3-,C2-和单C-类物质热分解的26个反应步骤,如图 4所示(方括号中的数据为速率分支比,圆括号中的字母为中间体或电子态的标记)。该图为1200K的势能剖面图。其中,C3-反应的决速步骤为:C3H3·(IM10)→C3H3·(IM11)→C3H2(IM13)+H·(·表示自由基),能垒为203.4kJ·mol-1和193.7kJ·mol-1;C2-和单C-类物质反应通道的高能垒为174.1kJ·mol-1和181.4kJ·mol-1。其中最高能垒203.4kJ·mol-1与实验[51]得到的表观活化能201.9kJ·mol-1±0.6kJ·mol-1符合很好。

Ge等[52]在其热力学研究[32]的基础上,进一步研究了MTS-H2热解制备SiC基体CVD气相反应体系中114个化学反应的动力学。发现其中73个反应有能垒(即TS)存在,用MP2得到了73个TS的结构,用CCSD(T)/aug-cc-pVTZ得到分子能量。在0~2000K内11个温度上,分别计算了形成73个TS的ΔrH、ΔrS和ΔrG。得到了在0K和1400K两个温度点CH3SiCl3,C2H6,C2H4,SiCl3+H2和SiCl2+H2等分解的5条重要反应通道。而对于自由基参与的反应,尚未考虑在其中。Wang等[53]采用DFT、G3(MP2)和统计热力学方法等理论方法,也详细研究了MTS在气相反应中最初的反应路径,其中包括了自由基进攻的反应步骤。根据所得到的298.15K和1200K的热力学数据进行分析,结果显示,MTS的直接分解,所需能量至少为339.8kJ·mol-1(298.15K)或210.1kJ·mol-1(1200K)。但如果以自由基进攻的方式分解,则能垒要明显降低,如图 5所示(右半图对应298.15K,左半图对应1200K)。以298.15K(右半图)为例,各种自由基进攻路径上的较低反应能垒,都在100kJ·mol-1以内。尤其是Cl自由基进攻MTS的一个H原子的能垒仅为41.2kJ·mol-1。这一工作,对于MTS的气相热分解反应的机理,给出了更合理的认识。
 |
图 5 自由基进攻MTS的初始分解路径[53] (a)CH3,CH2Cl,CH2Cl2和CCl3自由基进攻MTS;(b)H和Cl自由基进攻MTS;(c)SiCl3,SiHCl2,SiH2Cl和SiH3自由基进攻MTS Fig. 5 Initial pathways of decomposition of MTS attacked by free radicals[53] (a)MTS attacked by CH3,CH2Cl,CHCl2 and CCl3 radicals;(b)MTS attacked by H and Cl radicals;(c)MTS attacked by SiCl3,SiHCl2,SiH2Cl and SiH3 radicals |
Liu等[54]用DFT、G3(MP2)和统计热力学等理论方法,对CH4-BCl3-H2体系气相反应路径做了详细的研究与分析。结果显示,大部分有利的反应也都是自由基链反应。生成BC的最可能反应历程包含了9步反应,对应298.15K的较高能垒或能量为236.4,238.6kJ·mol-1和225.6 kJ·mol-1,对应1200K的较高能量为346.0,248.3,213.2kJ·mol-1和242.8kJ·mol-1。生成B的最可能反应历程包括5步反应,对应298.15K的较高的能垒或能量为224.6kJ·mol-1和294.7kJ·mol-1,对应1200K的能量为314.2,203.1kJ·mol-1和244.6kJ·mol-1。其中,1200K下生成BC最佳反应通道中最高能垒(346.0kJ·mol-1)与实验观测到的表观活化能的范围[55](152~473kJ·mol-1)也相当,对揭示不同反应阶段的控制步骤,提供了理论依据。进一步分析表明,在较低温度下,生成BC相对容易,而在较高温度下,生成B相对容易,这为正确认识该反应的机理,提供了理论依据。
类似地,Jiang等[56]用DFT、完全活性空间自洽场方法(CASSCF)、统计并结合经典热力学等方法,研究了BCl3-C3H6-H2体系热解的气相反应途径。对于用精确量子化学方法的研究而言,该体系较大、反应也更复杂。因此,作者采取了每一步分解反应穷尽所有可能(包括直接裂解和H、Cl自由基进攻反应),其后再选取能垒最低的步骤,以此步骤的产物作为下一级裂解的反应物。以此类推,最终得到体系裂解的“最低”反应通道。分析表明,绝大部分能量有利的反应通道也都对应自由基链反应,从而确定了体系热解的“最低”反应通道共有19个步骤。在298.15K下,生成C3B的决速步能垒为215.1kJ·mol-1。在1200K下时,最高能垒为275.1kJ·mol-1。决速步能垒数据(尤其是298.15K)与最新报道的[57]表观活化能实验数据208.7kJ/mol符合很好。
由于Jiang等[56]针对BCl3-C3H6-H2体系采用的方法,并不能保证找到真正的最佳反应通道,即所选择的上一步能垒最低的反应物,在后面的反应步骤中可能有较高的能垒,而能垒稍高一些的反应物在后续的反应过程中能垒则可能更低。因此,Yang等[58]进一步用同样的理论方法,研究了能垒次低(比最低通道能垒高28.5kJ·mol-1以内,或速率分支比大于10-5)的5条新通道,比较了得到的所有路径,最终确定出了最佳反应通道。结果表明,生成产物CB和C2B的最高能垒比生成C3B的要高,在298.15K下,生成C3B的最高能垒为192.1kJ·mol-1,相比Jiang的“最低”反应通道的215.1kJ·mol-1低了23kJ·mol-1;而在1200K下,最高能垒271.5kJ·mol-1比Jiang的275.1kJ·mol-1略低了3.6kJ·mol-1。这些工作表明,对于目前通过实验方法还很难开展的反应历程研究,量子力学方法具有独特的优势。
2.2 速率常数与反应速率为了与实验中可以测定的表观反应速率进行比较,同时也为了获得反应的动力学数学模型,在反应历程已知的情况下,还需要进一步确定各基元反应的速率常数,以及尝试导出表观速率常数或表观活化能的解析关系。目前,一般采用变分过渡态理论(VTST)方法计算基元反应的速率常数[59, 60],而表观速率常数与基元反应速率常数之间的解析关系,在复杂反应中并不一定存在。此时,也还需要借助其他数值方法求解耦合微分方程,获得各物质浓度与反应时间的关系,方可与实验结果进行比较。
Han等[61]对文献[50]得到的丙烯热解最佳反应通道的16个基元反应进行研究,借助于G3(MP2)获得的过渡态势能剖面特征参数; 用VTST确定了Arrhenius速率常数k1~k16;用稳态近似方法,推导出了丙烯热解正反应的表观(apparent)速率
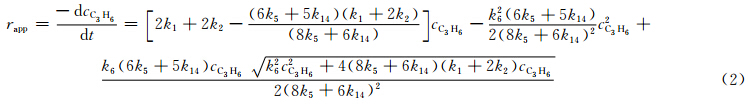
高温下,式(2)中后两项相对很小,因此可近似为简单的一级反应

虽然式(3)给出了该反应的表观速率和表观速率常数的解析关系,但并不存在表观活化能的表达式。根据式(3)和商业程序COMSOL用有限元数值方法求解相同的16个基元反应所对应的耦合微分方程的结果,反应物丙烯的浓度以及反应速率与时间的关系如图 6所示。图 6(a)表明高温下丙烯热解是一个快速反应,由式(3)和COMSOL(正反应)得到的丙烯浓度随时间的变化曲线基本重合;图 6(b)表明由式(3)和COMSOL(正反应)得到的反应速率曲线也非常接近。文献[61]还用一个典型实验温度1173.15K计算正反应丙烯浓度随时间的变化,结果表明丙烯被基本消耗完的时间0.55s,与最新实验的滞留时间0.6s[51]相符。用1200K的ΔrG导出的表观活化能215.7kJ·mol-1,也与最新报道的实验值201.9kJ·mol-1±0.6kJ·mol-1[51]符合很好,从而在理论上揭示了该体系的反应动力学特征。

对于文献[32, 52]所研究的化学反应,Ge等[62]采用过渡态理论(TST)等方法进一步研究了速率常数。结果显示,在不同温度和压力下,理论与已有实验比较,50余个反应速率常数与实验值偏差一般小于一个数量级,说明理论得到的数据是合理的。Liu等[63]针对文献[54]所得到CH4-BCl3-H2体系热解的最佳反应通道,进一步用VTST方法计算了最佳反应通道中16个基元反应在200~2000K的速率常数,用数值方法(COMSOL程序),获得了1200~1600 K温度和一定初始浓度下,各反应物浓度随时间的变化如图 7所示。图 7显示,随着温度的升高,CH4和BCl3的消耗速率均迅速加快。两种完全解离产物BC和B应是形成碳化硼的气相组元,即气相成核和表面沉积反应的重要物种,其浓度变化如图 8所示。由图 8可见,气相中生成BC和B的浓度都比较低,但随着温度的升高,浓度会显著增加;在1200~1600K温度范围内,B的浓度总是高于BC,因此在较高温度范围内可得富硼的碳化硼产物。利用CH4消耗速率与温度的关系,还得到了该反应的表观活化能为208.4kJ·mol-1;而以BCl3的速率来衡量,反应在高温和较低温度下,分别遵循不同的机制,对应的表观活化能分别为378.3kJ·mol-1和201.7kJ·mol-1。这些数据与G3(MP2)计算的决速步垒238.6kJ·mol-1和文献[55, 64]中给出的表观活化能范围152.87~473kJ·mol-1都可比拟。


对于较为复杂的BCl3-C3H6-H2体系,Xiao等[65]在最佳反应通道[56, 58]的基础上,用VTST方法确定了各基元反应的速率常数,仍然用COMSOL有限元数值方法,求解了化学反应速率,获得了它们与温度的关系。在典型的实验条件下,获得了1100~1500K反应物和主要产物浓度随时间的关系。结果显示,在1100,1200,1300,1400K和1500K五个温度下,丙烯几乎完全消耗的反应时间分别为30,1.5,0.12,0.011s和0.001s;反应产物虽有C3B、C2B和CB,但在高温下C3B是主要产物,在较低温度下C2B是主要产物。在反应经过1s时,温度在1100~1500K以内,产物浓度都能接近平衡,但浓度都较低。以反应物丙烯的消耗速率计,得到反应的表观活化能为337.0kJ·mol-1,比实验值[66]208.7kJ·mol-1大,但与决速步势垒362.6kJ·mol-1符合很好。该表观活化能与实验值存在的差别,很可能需要进一步的研究。
3 结束语先驱体热解制备陶瓷材料的反应机理,有时虽然异常复杂,用实验方法很难开展研究。但用基于第一性原理的理论方法,已可以获得各种气态小分子的结构和精确能量,结合统计热力学方法,获得其任意温度的热化学数据,实现系统性地研究反应热力学规律和反应途径、反应速率等动力学问题,并在C3H6(丙烯)+H2,MTS+H2,CH4+BCl3+H2,C3H6(丙烯)+BCl3+H2体系的研究中得到应用,为它们热解制备层状碳、抗氧化SiC以及自愈合BxC陶瓷的成分控制和工艺优化提供了科学基础。
但这一课题仍然存在一些问题尚需深入研究。例如:(1) 反应系统中往往同时产生多种高活性中间体(自由基、激发态、离子态物质),而这些中间体还可相互反应或相互结合,形成更大的分子。这些分子的存在对材料制造有何影响,还缺乏系统性的研究。(2) 作为CVD/CVI制备材料的全过程,除了先驱体的气相分解之外,还有如关键物种的气相成核、气相扩散、表面扩散、表面沉积和表面反应等环节。目前的材料制造机理研究中,这些环节还有待深入。(3) 在理论方法方面,一般的稳定分子能量的精确计算方法是经过严格检验的。但如自由基、激发态、离子态物质以及非稳定结构的过渡态,在其电子结构方面,很可能还存在显著的非动态电子相关(Non-dynamic electronic correlations)效应。目前的理论方法,还没有系统性地做过精度检验,也还没有建立起一套普适的可接受的[67]精确算法。
| [1] | SCHMIDT S, BEYER S, KNABE H, et al. Advanced ceramic matrix composite materials for current and future propulsion technology applications[J].Acta Astronautica,2003,55(3):409-420. |
| [2] | NASL R. Design, preparation and properties of non-oxide CMCS for application in engines and nuclear reactors: An overview: Symposium on multifunctional materials and structures[J]. Composites Science and Technology, 2003, 64(2): 155-170. |
| [3] | 王锟, 陈刘定, 郑翔. 平纹编制C/SiC复合材料在室温和高温环境下的拉伸行为[J]. 航空材料学报, 2010, 30(1): 78-84. WANG Kun, CHEN Liu-ding, ZHENG Xiang. Comparison of tensile behavior of plain-woven carbon/silicon carbide composites at room temperature and high temperature[J]. Journal of Aeronautical Materials, 2010, 30(1): 78-84. |
| [4] | XU Y D, CHENG L F, ZHANG L T, et al. Carbon silicon carbide composites prepared by chemical vapor infiltration combined with silicon melt infiltration[J].Carbon,1999,37(8):1179-1187. |
| [5] | QUEMARDA L, REBILLAT F, GUETTE A, et al. Self-healing mechanisms of a SiC fiber reinforced multi-layered ceramic matrix composite in high pressure steam environments[J]. Journal of the European Ceramic Society, 2007, 27(4): 2085-2094. |
| [6] | 周新贵, 张长瑞, 何新波, 等. 热解碳涂层碳纤维增强碳化硅复合材料热压工艺研究[J]. 材料工程, 2000, (3): 39-41. ZHOU Xin-gui, ZHANG Chang-rui, HE Xin-bo, et al. A study on the hot press of pyrolysised carbon coated carbon fiber reinforced silicon carbide composite[J]. Journal of Materials Engineering, 2000, (3): 39-41. |
| [7] | 焦桓, 周万城, 李翔. CVD法水蒸气条件下制备SiC块体[J]. 材料工程, 2000, (12): 12-14,18. JIAO Huan, ZHOU Wan-cheng, LI Xiang. Growth rate and deposition process of SiC with water vapor introduced by CVD[J]. Journal of Materials Engineering, 2000, (12): 12-14,18. |
| [8] | WU S J, CHENG L F, YANG W B, et al. Oxidation protective multilayer CVD SiC coatings modified by a graphitic B-C interlayer for 3D C/SiC composite[J]. Applied Composite Materials, 2006, 13(6): 397-406. |
| [9] | FARIZY G, CHERMANT J L, SANGLEBOEUF J C, et al. SiCf-SiBC composites: microstructural investigations of the as received material and creep tested composites under an oxidative environment[J]. Journal of Microscopy-oxford, 2003, 210(2): 176-186. |
| [10] | 张立同, 成来飞, 徐永东, 等. 自愈合碳化硅陶瓷基复合材料研究及应用进展[J]. 航空材料学报, 2006, 26(3): 226-232. ZHANG Li-tong, CHENG Lai-fei, XU Yong-dong, et al. Progress on self-healing silicon carbide ceramic matrix composites and its applications[J]. Journal of Aeronautical Materials, 2006, 26(3): 226-232. |
| [11] | POPLE J A, HEAD-GORDON M, FOX D J, et al. Gaussian-1 theory: a general procedure for prediction of molecular energies[J]. The Journal of Chemical Physics, 1989, 90(10): 5622-5629. |
| [12] | CURTISS L A, JONES C, TRUCKS G W, et al. Gaussian-1 theory of molecular energies for second-row compounds[J]. The Journal of Chemical Physics, 1990, 93(4): 2537-2545. |
| [13] | CURTISS L A, RAGHAVACHARI K, TRUCKS G W, et al. Gaussian-2 theory for molecular energies of first-and second-row compounds[J]. The Journal of Chemical Physics, 1991, 94(11): 7221-7230. |
| [14] | CURTISS L A, RAGHAVACHARI K, POPLE J A. Gaussian-2 theory using reduced møller-pleset orders[J]. The Journal of Chemical Physics, 1993, 98(2): 1293-1298. |
| [15] | CURTISS L A, CARPENTER J E, RAGHAVACHARI K, et al. Validity of additivity approximations used in Gaussian-2 theory[J]. The Journal of Chemical Physics, 1992, 96(12): 9030-9034. |
| [16] | CURTISS L A, RAGHAVACHARI K, REDFERN P C, et al. Gaussian-3 theory for molecules congaining first and second-row atoms[J]. 1998, 109(18): 7746-7754. |
| [17] | CURTISS L A, REDFERN P C, RAGHAVACHARI K, et al. Gaussian-3 theory using reduced Møller-Pleset order[J]. The Journal of Chemical Physics, 1999, 110(10): 4703-4709. |
| [18] | BABOUL A G, CURTISS L A, REDFERN P C, et al. Gaussian-3 theory using density functional geometries and zero-point energies[J]. The Journal of Chemical Physics, 1999, 110(10): 7650-7657. |
| [19] | CURTISS L A, REDFERN P C, RAGHAVACHARI K, et al. Assessment of gaussian-3 and density-functional theories on the G3/05 test set of experimental energies[J]. The Journal of Chemical Physics, 2005, 123(12): 124107-1-12. |
| [20] | CURTISS L A, REDFERN P C, KRISHNAN R. Gaussian-4 theory[J]. The Journal of Chemical Physics, 2007, 126(8): 084108-1-12. |
| [21] | CURTISS L A, REDFERN P C, KRISHNAN R. Gaussian-4 theory using reduced order perturbation theory[J]. The Journal of Chemical Physics, 2007, 127(12): 124105-1-8. |
| [22] | NYDEN M R, PETERSSON G A. Complete basis set correlation energies.1.the asymptotic convergence of pair natural orbital expansions[J]. The Journal of Chemical Physics, 1981, 75(4):1843-1862. |
| [23] | PETERSSON G A, BENNETT A, TENSFELDT T G, et al. A complete basis set model chemistry.1.The total energy of closed-shell atoms and hydrides of the first-row elements [J]. The Journal of Chemical Physics, 1988, 89(4): 2193-2218. |
| [24] | PETERSSON G A, ALLAHAM M A. A complete basis set model chemistry.2.Open-shell systems and the total energies of the first-row atoms[J]. The Journal of Chemical Physics, 1991, 94(9): 6081-6090. |
| [25] | PETERSSON G A, TENSFELDT T G, MONTGOMERY J A, et al. A complete basis set model chemistry.3.The complete basis set-quadratic configuration interaction family of methods[J]. The Journal of Chemical Physics, 1991, 94(9): 6091-6101. |
| [26] | MONTGOMERY J A, OCHTERSKI J W, PETERSSON G A. A complete basis set model chemistry.4.an improved atomic pair natural orbital method[J]. The Journal of Chemical Physics, 1994, 101(7): 5900-5909. |
| [27] | OCHTERSKI J W, PETERSSON G A, MONTGOMERY J A. A complete basis set model chemistry.5.Extensions to six or more heavy atoms[J]. The Journal of Chemical Physics, 1996, 104(7):2598-2619. |
| [28] | MONTGOMERY J A, FRISCH M J, OCHTERSKI J W, et al. A complete basis set model chemistry.6.Use of density functional geometries and frequencies[J]. The Journal of Chemical Physics, 1999, 110(6): 2822-2827. |
| [29] | WOOD G P F, PETERSSON G A, BARNES E C, et al. A restricted-open-shell complete-basis-set model chemistry[J]. The Journal of Chemical Physics, 2006, 125(9):094106-1-16. |
| [30] | BARNES E C, PETERSSON P A. MP2/CBS atomic and molecular benchmarks for H through Ar[J]. The Journal of Chemical Physics, 2010, 132(11): 114111-1-9. |
| [31] | DUMINDA S R, PETERSSON P A. CCSD(T)/CBS atomic and molecular benchmarks for H through Ar[J]. The Journal of Chemical Physics, 2013, 138(14): 144103-1-12. |
| [32] | GE Y, GORDON M S, BATTAGLIA F, et al. Theoretical study of the pyrolysis of methyltrichlorosilane in the gas phase.1.thermodynamics[J]. Journal of Physical Chemistry A, 2007, 111(8): 1462-1474. |
| [33] | DENG J L, SU K H, ZENG Y, et al. Investigation of thermodynamic properties of gaseous SiC(X3Π and a1Σ) with accurate model chemistry calculations[J]. Physical A: Statistical Mechanics and Its Applications, 2008, 387(22): 5440-5456. |
| [34] | YAO X P, SU K H, DENG J L, et al. Gas-phase reaction thermodynamics in preparation of pyrolytic carbon by propylene pyrolysis[J]. Computational Materials Science, 2007, 40(4): 504-524. |
| [35] | DENG J L, SU K H, YAO X P, et al. Erratum to: ''Gas-phase reaction thermodynamics in preparation of pyrolytic carbon by propylene pyrolysis[J]. Computational Materials Science, 2008, 44(2): 838-840. |
| [36] | DAVID R L. CRC Handbook of Chemistry and Physics, 77th ed[M]. New York: CRC Press, 1996-1997. |
| [37] | CHASE M W, DAVIES C A, DOWNEY J R, et al. NIST-JANAF Thermochemical Tables Forth Edition [M]. New York: American Chemical Society and American Institute of Physics, 1998. |
| [38] | DENG J L, SU K H, WANG X, et al. Thermodynamics of the gas-phase reactions in the chemical vapor deposition of silicon-carbide with methyltrichlorosilane precursor[J]. Theoretical Chemistry Account, 2009, 122(1-2): 1-22. |
| [39] | DENG J L, SU K H, WANG X, et al. Thermodynamics of the production of condensed phases in the chemical vapor deposition process of methyltrichlorosilane pyrolysis[J]. Chemical Vapor Deposition, 2009, 15(10-15): 281-290. |
| [40] | ZENG Q F, SU K H, ZHANG L T, et al. Evaluation of the thermodynamic date of CH3SiCl3 based on quantum chemistry calculations[J]. Journal of Physical and Chemical Reference Date, 2006, 35(2): 1385-1390. |
| [41] | ZENG Y, SU K H, DENG J L, et al. Thermodynamic investigation of the gas-phase reactions in the chemical vapor deposition of boron carbide with BCl3-CH4-H2 precursors[J]. Journal of Molecular Structure Theochem, 2008, 861(1-3): 103-116. |
| [42] | WANG T, SU K H, DENG J L, et al. Reaction thermodynamic in chemical vapor deposition of boron carbide with BCl3-C3H6-H2 precursors[J]. Journal of Theoretical & Computational Chemistry, 2008, 7(6): 1269-1312. |
| [43] | 邓娟利. CVD/CVI制备自愈合SiC陶瓷基复合材料的反应热力学研究[D]. 西安: 西北工业大学, 2009. DENG Juan-li. Thermodynamics of the reaction in the CVD/CVI preparation of the self-healing SiC ceramic matrix composite materials[D]. Xi'an: Northwestern Polytechnical University, 2009. |
| [44] | DENG J L, CHENG L F, ZHANG L T, et al. Thermodynamics of the production of condensed phases in the chemical vapor deposition process of zirconium diboride with ZrCl4-BCl3-H2 precursors[J]. Thin Solid Films, 2012, 520(6): 2331-2335. |
| [45] | DENG J L, CHENG L F, ZHANG L T, et al. Thermodynamic on study co-deposition of ZrB2-SiC from ZrCl4-BCl3-CH3SiCl3-H2 system[J]. Thin Solid Films, 2012, 520(23): 7030-7034. |
| [46] | DENG J L, CHENG L F, ZHENG G P, et al. Thermodynamics on formation of condensed phases during CVD Si3N4 process with SiCl4-NH3-H2 precursors[J]. Advanced Engineering Materials, 2011, 194-196: 1516-1523. |
| [47] | XUE J M, YIN X W, YE F, et al. Thermodynamic analysis on the codeposition of SiC-Si3N4 composite ceramics by chemical vapor deposition using SiCl4-NH3-CH4-H2-Ar mixture gases[J]. Journal of the American Ceramic Society,2013,96(3):979-986. |
| [48] | LIU Q F, ZHANG L T, LIU J, et al. Thermodynamic study on codeposition of ZrC-SiC from MTS-ZrCl4-CH4-H2[J].Inorganic Materials, 2010, 46(10): 1090-1095. |
| [49] | LIU X F, ZHANG L T, LIU Y S, et al. Thermodynamic calculations on the chemical vapor deposition of Si-C-N from the SiCl4-NH3-C3H6-H2-Ar system[J]. Ceramics International, 2013, 39(4): 3971-3977. |
| [50] | QU Y N, SU K H, WANG X, et al. Reaction pathways of propene pyrolysis[J]. Journal of Computational Chemistry, 2009, 31(7): 1421-1442. |
| [51] | 赵春年, 成飞来, 张立同, 等. 丙烯化学气相沉积热解碳的动力学研究[J]. 无机材料学报, 2008, 23(6): 1165-1170. ZHAO Chun-nian, CHENG Lai-fei, ZHANG Li-tong, et al. In-suit kinetics study in chemical vapor deposition of pyrocarbon from propylene[J]. Journal of Inorganic Materials, 2008, 23(6): 1165-1170. |
| [52] | GE Y, GORDON M S, BATTAGLIA F, et al. Theoretical study of the pyrolysis of methyltrichlorosilane in the gas phase.2.reaction paths and transition states[J]. Journal of Physical Chemistry A, 2007, 111(8): 1475-1486. |
| [53] | WANG X, SU K H, DENG J L, et al. Initial decomposition of methyltrichlorosilane in the chemical vapor deposition of silicon-carbide[J]. Computational and Theoretical Chemistry, 2011, 967(2-3): 265-272. |
| [54] | LIU Y, SU K H, ZENG Q F, et al. Reaction paths of BCl3 + CH4 + H2 in the chemical vapor deposition process[J]. Structural Chemistry, 2012, 23(6): 1677-1692. |
| [55] | BERJONNEAU J, LANGLAIS F, CHOLLON G, et al. Understanding the CVD process of (Si)-B-C ceramics through FTIR spectroscopy gas phase analysis[J]. Surface and Coatings Technology, 2007, 201(16-17): 7273-7285. |
| [56] | JIANG X Q, SU K H, WANG X, et al. An investigation of the lowest reaction pathway of propene+BCl3 decomposition in chemical vapor deposition process[J]. Theoretical Chemistry Accounts, 2010, 127(5-6): 519-538. |
| [57] | LIU Y S, ZHANG L T, CHENG L F, et al. Uniform design and regression analysis of LPCVD boron carbide from BCl3-CH4-H2 system[J]. Applied Surface Science, 2009, 255(11): 5729-5735. |
| [58] | YANG J H, SU K H, LIU Y, et al. New reaction paths of propene + BCl3 decomposition in chemical vapor deposition process[J]. Journal of Theoretical & Computational Chemistry, 2012, 11(1): 53-85. |
| [59] | TRUHLAR D G, GARRETT B C. Variational transition state theory[J]. Annual Review of Physical Chemistry, 1984, 35: 159-189. |
| [60] | GONZALES-LAFONT A, TRUONG T N, TRUHLAR D G. Interpolated variational transition-state theory: practical methods for estimating variational transition-state properties and tunneling contributions to chemical reaction rates from electronic structure calculations[J]. The Journal of Chemical Physics, 1991, 95(12): 8875-8894. |
| [61] | HAN P P, SU K H, WANG Y L, et al. Reaction rate of propene pyrolysis[J]. Journal of Computational Chemistry, 2011, 32(13): 2745-2755. |
| [62] | GE Y, GORDON M S, BATTAGLIA F, et al. Theoretical study of the pyrolysis of methyltrichlorosilane in the gas phase.3.reaction rate constant calculations[J]. Journal of Physical Chemistry A, 2010, 114(6): 2384-2392. |
| [63] | 刘艳. H2O-SiC(001)及CH4-BCl3-H2反应动力学研究[D]. 西安: 西北工业大学, 2012. LIU Yan. Reaction kinetics of H2O-SiC(001) and CH4-BCl3-H2 decomposition system[D]. Xi'an: Northwestern Polytechnical University, 2012. |
| [64] | YE F E, ZHANG L T, CHENG L F, et al. Effect of temperature on deposition process of boron doped carbon from BCI3-CH4-H2 by chemical vapor deposition[J]. Materials Review, 2010, 24(7): 108-115. |
| [65] | XIAO J, SU K H, LIU Y, et al. Decomposition reaction rate of BCl3-C3H6-H2 in gas phase[J]. Journal of Physical Chemistry A, 2012, 116(26): 6955-6966. |
| [66] | LIU Y S, ZHANG L T, CHENG Y S, et al. Preparation and mechanical properties of carbon fiber reinforced (BCx-SiC)(n) multilayered matrix composites[J]. Applied Composite Materials, 2007, 14(4): 277-286. |
| [67] | SΦLLING T I, SMITH D M, RADOM L, et al, Towards multireference equivalents of the G2 and G3 methods[J]. Journal of Chemical Physics, 2001, 115(19): 8758-8772. |