 2021, Vol. 49
2021, Vol. 49文章信息
- 黄居峰, 宋光铃
- HUANG Ju-feng, SONG Guang-ling
- 镁合金阳极的析氢、效率与电偶腐蚀放大效应
- Hydrogen evolution, efficiency and exacerbated galvanic corrosion damage of magnesium alloy anode
- 材料工程, 2021, 49(12): 48-56
- Journal of Materials Engineering, 2021, 49(12): 48-56.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2021.000287
-
文章历史
- 收稿日期: 2021-03-31
- 修订日期: 2021-05-12




2. 厦门大学 固体表面物理化学国家重点实验室, 福建 厦门 361005;
3. 昆士兰大学 机械和采矿学院, 澳大利亚 布里斯班 4072
2. State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen University, Xiamen 361005, Fujian, China;
3. School of Mechanical and Mining Engineering, The University of Queensland, Brisbane 4072, Australia
镁合金具有优异的力学性能和化学性质,作为结构材料、生物材料以及电池材料得到广泛研究。由于镁的平衡电位很负(-2.37 V vs SHE),实际工程应用中当与其他工程金属耦接时镁合金都将作为电偶阳极发生腐蚀。据此,镁合金也可以作为牺牲阳极保护其他工程金属的正常服役[1-2]。除此之外,镁合金还可以作为镁空气电池的阳极材料[3-4],通过调控镁合金的成分和结构来提高镁合金的阳极效率和放电性能。可见,不管是作为工程结构金属,还是像牺牲阳极和电池电极那样的功能材料,深入理解镁合金的阳极溶解反应是保证其实际有效应用的基础。
镁合金的电偶腐蚀不同于它的自然腐蚀,由于镁合金的自腐蚀电位受高纯化与合金化影响较小,高纯化和合金化的方式很难改变镁合金被耦接金属加速溶解的电偶阳极过程,因此通过提高耐自腐蚀的方法很难有效抑制镁合金的电偶腐蚀。而实际工程应用中镁合金无法避免与其他金属接触,因此镁合金的电偶腐蚀才是镁合金工程应用的真正瓶颈。
在电偶腐蚀中,镁合金阳极溶解的本质是镁合金被耦合阴极相阳极极化而加速溶解的问题,阳极化的镁合金会产生负差数效应。镁合金的负差数效应是指阳极化的镁合金实际溶解速率“异常”地高于法拉第计算值,并伴随着析氢速率随着阳极电位或电流的增大而增大的“反常现象”[5-6]。因此,负差数效应导致的异常加速镁阳极溶解过程,放大了电偶腐蚀。研究发现,镁合金/铁电偶上根据电偶理论通过计算机模拟计算得到理论电偶腐蚀深度与实际测量的电偶腐蚀深度相比,实际的腐蚀深度远高于电偶效应预测的深度[7]。另外,镁的阳极溶解过程中产生的表面碱化、合金元素的溶解以及众多中间反应产物,再加上电偶腐蚀中阴极相的溶解和环境离子加入,各种产物的相互作用给镁合金的电偶腐蚀带来未知的“次生效应”。例如,镁合金腐蚀后产生氢氧根能够影响铝合金和锌的腐蚀,可以减轻电偶腐蚀;而碳钢腐蚀溶解后的铁离子转移到镁合金表面将加速镁合金的腐蚀,产生“毒化”作用;除此之外,电偶间的腐蚀产物还会产生“短路”效应,导致电偶远端发生意想不到的局部腐蚀[8]。可见,镁合金的“负差数效应”和“次生效应”使得实际镁合金的电偶腐蚀在程度和分布上变得复杂和难以预测。
不论这些腐蚀现象多么复杂,其核心的科学问题是镁溶解过程中伴随的析氢机制。对阳极析氢和阴极析氢机理的不同认识,决定着不同镁合金腐蚀防护策略的采用,最终将导致防腐效果的巨大差异。本工作测量了镁合金的阴阳极析氢速率,旨在厘清镁合金阳极溶解过程与析氢机理;通过理论推导得到镁阳极溶解放大倍数的主要影响因素,解释了镁阳极效率与腐蚀的关系,指导镁合金和镁阳极的制备与应用。
1 实验材料与方法实验材料为铸态纯镁、铸态AZ91镁合金以及镁丝(ϕ=0.2 mm),其化学成分如表 1所示。纯镁和镁合金切割成尺寸为1 cm×1 cm×1 cm的试样,采用环氧树脂进行封装制成电极。镁丝阵列电极是由8根同一方向排列的镁丝封装在环氧树脂中制备而成,其中每根镁丝用细铜线引出。实验配制了9种溶液,分别为0.01,0.1 mol/L和0.2 mol/L的盐酸溶液,0.05 mol/L的硫酸溶液,饱和氢氧化镁溶液,0.02,0.2 mol/L和2 mol/L的氯化钠溶液以及5 mmol/L的氯化锌溶液,其中氯化钠溶液和氯化锌溶液的pH值用稀盐酸调节为6,且氯化锌溶液中的氯离子浓度用氯化钠粉末调节到0.02 mol/L。
| Electrode material | Al | Zn | Fe | Cu | Ni | Zr | Mg |
| Pure Mg | 0.1066 | 0.0046 | 0.0022 | < 0.0006 | < 0.0001 | < 0.0001 | Bal |
| AZ91 alloy | 9.101 | 0.8802 | 0.0011 | 0.0007 | 0.00005 | Bal | |
| Mg wire | 0.0512 | 0.0149 | 0.008 | 0.0013 | 0.0053 | Bal |
采用Gamry Reference 600+电化学工作站对封装后的纯镁电极、AZ91镁合金电极以及镁丝阵列电极在不同溶液中进行恒电流极化,并采用排水法利用烧杯-漏斗-滴定管装置进行氢气体积的实时测量[9]。在室温下,电化学实验采用三电极体系,上述3种电极为工作电极,铂片为辅助电极,饱和氯化银电极为参比电极,500 mL的上述溶液为腐蚀介质。在恒电流测试中,首先在开路条件下稳定10 min,然后进行不同电流密度的阳极极化实验,电化学工作站记录电位随时间变化曲线,并且每隔一段时间人工读取记录析氢体积,根据式(1)可以计算得出析氢电流密度IH。
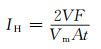
|
(1) |
式中:V表示极化时间t内气体的体积;F为法拉第常数;Vm为气体摩尔体积;A为镁电极的暴露面积。
镁丝阵列电极的极化实验的原理如图 1所示,采用多通道零电阻电流计(ZRA)记录流经每根镁丝的电流变化值。采用DVM-6a型徕卡光学显微镜原位观察阳极极化纯镁电极的表面变化。采用LEO-1530型扫描电子显微镜(SEM)观察阳极极化后的纯镁截面形貌。

|
图 1 镁丝阵列电极极化实验示意图 Fig. 1 Schematic diagram of polarization experiment of magnesium wire array electrode |
为了讨论纯镁的析氢反应和阳极效率变化,本课题组分别在0.01,0.1 mol/L和0.2 mol/L盐酸溶液中对纯镁电极进行恒电流阳极极化。图 2为不同浓度盐酸溶液中纯镁的测量电位、析氢电流和阳极效率随外加阳极极化电流的变化图。由图可知,析氢电流随着阳极极化电流的增大先减小后增大,最低析氢电流随着溶液酸度的增加而增大,对应的测量电位在-1.0 V附近。在不同极化阳极电位区间(-1.0 V前后)两种不同析氢速率的变化趋势表明存在两种不同的析氢过程:阳极析氢和阴极析氢。正常的阴极析氢可以根据Butler-Volmer方程描述,随着阳极电位或电流的增大,析氢速率减小;在析氢平衡电位附近,析氢停止。而阳极析氢是“负差数效应”的一种表观现象,其速率随阳极极化的增大而加速。显然随着阳极电流的增大,镁表面的阴极反应被抑制,阴极析氢电流密度减小。随着阳极极化的继续增强,更多的单价镁离子产生,阳极析氢电流密度增大。图 2还展示了纯镁的阳极效率随着阳极极化电流的变化趋势。当阳极极化电流密度小于析氢最低点对应的阳极电流密度时,随着极化电流密度的增大,阳极效率增大;在酸性较大的溶液中,纯镁的阳极效率保持上升,后趋近于63%。

|
图 2 在不同浓度盐酸溶液中纯镁的测量电位、析氢电流和阳极效率随外加阳极极化电流的变化图(极化时间为2 h) Fig. 2 Measured potential, hydrogen evolution current and anode efficiency of pure magnesium at different galvanostatic anodic current densities in different concentrations of hydrochloric acid solutions(the polarization time is 2 h) |
图 3与图 4分别为阳极极化纯镁的腐蚀形貌的光学和SEM图。外加阳极极化电流为10 mA/cm2时,析氢过程是由阴极析氢控制,电极表面具有光滑的金属色(图 3(a))。外加阳极电流密度为20 mA/cm2时,析氢电流达到了最小值,且此时纯镁表面恰好出现黑色腐蚀产物(图 3(b))。外加阳极极化电流为50 mA/cm2时,析氢过程是由阳极析氢控制部分,由于镁的阳极溶解,其表面黑色腐蚀产物增多(图 3(c)),且随阳极极化的增强,腐蚀变得更加不均匀(图 4),这与文献[10]的报道是一致的。另外,溶液的酸度越大,表面的覆盖度越小,不管是阴极析氢还是阳极析氢,在裸露的金属镁表面上都会变得容易进行。

|
图 3 在0.1 mol/L的盐酸溶液中不同阳极电流极化后纯镁的光学图片 (a)10 mA/cm2; (b)20 mA/cm2; (c)50 mA/cm2 Fig. 3 Optical images of anodized pure Mg in 0.1 mol/L hydrochloric acid solution (a)10 mA/cm2; (b)20 mA/cm2; (c)50 mA/cm2 |

|
图 4 在0.1 mol/L盐酸溶液中不同阳极电流极化后纯镁的表面截面SEM图 (a)30 mA/cm2; (b)60 mA/cm2 Fig. 4 Cross-sectional SEM images of pure Mg after polarization in 0.1 mol/L hydrochloric acid (a)30 mA/cm2; (b) 60mA/cm2 |
上述盐酸溶液作为强腐蚀溶液,膜覆盖度对负差数效应的影响较小。但对于中性或碱性溶液,如氯化钠、硫酸钠以及氢氧化镁溶液,阳极极化条件下的“负差数效应”受表面膜的完整性影响较大[11]。图 5展示了不同浓度氯化钠溶液中的析氢电流和阳极效率变化图。纯镁在氯化钠溶液中具有明显的负差数效应,阳极析氢电流受氯离子的浓度影响较小。开路电位极化电流为零时,阳极效率只能为零;阳极电流较小时,因实验误差,第一个点的阳极效率偏高;阳极电流较大时,阳极效率基本稳定在59%左右。

|
图 5 在不同浓度氯化钠溶液中纯镁的析氢电流和阳极效率随外加阳极极化电流的变化(极化时间为2 h) Fig. 5 Hydrogen evolution current and anode efficiency of pure magnesium at different galvanostatic anodic current densities in different concentrations of sodium chloride solution (the polarization time is 2 h) |
实际的镁表面是不均匀的,阳极极化时镁表面有可能在某些局部区域出现阴极行为。这种表面不均匀导致的微电偶有可能提高阴极析氢量而降低阳极效率,从而使宏观电偶腐蚀被进一步放大。因此,本研究采用镁丝阵列电极模拟镁表面不同区域发生的腐蚀反应。镁丝阵列电极分别置于饱和氢氧化镁溶液和2 mol/L的氯化钠溶液中恒电流阳极极化,并利用多通道零电阻电流计检测不同镁丝的电流大小和极性。图 6展示了不同阳极极化电流下镁丝阵列电极上的电流密度分布以及总的阴阳极电流。图 6(a-1),(a-2)中不同颜色的立柱代表阵列电极中各个细丝电极的电流密度。在饱和氢氧化镁溶液中,当外加阳极极化电流为0.2 mA/cm2时,镁丝阵列电极中有的镁丝流经阳极电流,有的镁丝流经阴极电流;当外加阳极电流为0.5 mA/cm2和1 mA/cm2时,镁丝阵列电极中全部为阳极电流。另外,随着阳极极化的增强,每根镁丝之间的差别变小,总阴极电流消失,而总阳极电流逐渐增加。在2 mol/L的氯化钠溶液中,当外加阳极极化电流为2,6 mA/cm2和10 mA/cm2时,镁丝阵列电极中同时存在着阳极电流和阴极电流,只是流经阴阳极电流的镁丝数量不同;当外加阳极电流为20 mA/cm2时,镁丝阵列电极中全部为阳极电流。随着外加阳极电流密度的增加,镁丝阵列电极上流经阴极电流的镁丝数量减少,总阴极电流逐渐减小,而总阳极电流逐渐增加。镁丝中流经不同极性的电流,代表着表面发生阴阳极反应,不同镁丝之间形成微电偶。随着外加阳极电流密度增大,微电偶的作用减少。

|
图 6 在饱和氢氧化镁溶液(1)和2 mol/L的氯化钠溶液(2)中不同阳极电流极化下镁丝阵列电极上的电流密度分布图(a) 以及总的阴阳极电流和总电流图(b)(极化时间为2 h) Fig. 6 Current density distribution mapping (a), total anodic, cathodic and total current (b) of the magnesium wire array electrode under different anodic current polarization in saturated magnesium hydroxide solution(1) and 2 mol/L sodium chloride solution(2) (the polarization time is 2 h) |
AZ系列镁合金是常见的镁合金类型,铝元素的添加可增强这类镁合金的力学性能。另外在镁合金表面形成的氧化铝提高了镁合金的耐蚀性。图 7为纯镁和AZ91镁合金在0.1 mol/L盐酸溶液中析氢电流密度和阳极效率随外加阳极极化电流的变化图。图 7显示了AZ91镁合金的析氢电流密度随外加阳极电流密度的变化趋势与纯镁是一样的,存在明显的“负差数效应”。在相同的外加阳极极化电流下,AZ91镁合金的析氢电流密度明显高于纯镁,阳极效率低于纯镁。

|
图 7 纯镁和AZ91镁合金在0.1 mol/L盐酸溶液中析氢电流密度和阳极效率随阳极极化电流的变化(极化时间为2 h) Fig. 7 Hydrogen evolution current density and anode efficiency of pure magnesium and AZ91 magnesium alloy under different galvanostatic anodic current densities in 0.1 mol/L HCl solution (the polarization time is 2 h) |
镁合金都是多晶金属,除了基体相,还含有其他相。自腐蚀条件下,其他相作为局部阴极相与镁基体构成微电偶。β-Mg17Al12相是AZ系列镁合金中第二相,对阳极析氢有一定的影响;Mg17Al12相本身也具有负差数效应,当阳极极化电位大于Mg17Al12相的击穿电位时,Mg17Al12相的钝化状态被打破,也会发生阳极溶解和阳极析氢[12]。
2.4 溶液中离子的影响镁基体、合金元素溶解到腐蚀溶液中后,与溶液中的某些成分反应,进而影响腐蚀过程,将会给镁的电偶腐蚀带来“次生效应”的影响。
纯镁分别在0.1 mol/L盐酸和0.05 mol/L的硫酸溶液中进行恒电流阳极极化,控制溶液的酸度相同。图 8为不同阴离子对不同阳极极化电流下析氢电流和阳极效率的影响图。由图可知,不同酸性溶液中析氢电流随极化电流的变化趋势相同,硫酸溶液中镁的阳极效率随阳极极化电流密度的趋势与盐酸溶液中也相同,说明氯离子并不是导致析氢速率“异常”增加的原因,这与之前的研究结果是一致的[13]。但相对于硫酸溶液,氯离子的存在促进了Mg在阳极极化下的溶解,使得阳极析氢速率加快,这可能是由于氯离子比硫酸根离子对镁的腐蚀性更强[11]。

|
图 8 不同阴离子对不同阳极极化电流密度下析氢电流和阳极效率的影响(极化时间为2 h) Fig. 8 Effect of different anions on the hydrogen evolution density and anode efficiency under different polarization current densities (the polarization time is 2 h) |
图 9为在氯化钠和氯化锌溶液中的析氢电流密度和阳极效率随极化电流的变化图。由图可知,析氢电流随着外加阳极极化电流增大而增大,表现出明显的“负差数效应”。值得注意的是,在小的阳极极化电流(0.5 mA/cm2和1 mA/cm2)时,在氯化锌溶液中纯镁的析氢速率大于在氯化钠溶液中,但是在大的阳极极化电流(2 mA/cm2和5 mA/cm2)时,两种溶液中纯镁的析氢速率接近相等。在氯化钠溶液中,当阳极化较弱时,由于析氢量少,实验误差大,如前所述,导致阳极效率比实际偏大。随着外加阳极电流的增大,误差减少,阳极效率回归稳定值。而在含氯化锌溶液中,就算极化电流较小时,由于析氢量较大,误差小,呈现出正常的阳极效率随着外加阳极电流的增大而增大的现象。两种溶液中阳极效率最后都稳定在58%左右。由于锌离子的存在,可加速镁的腐蚀[14]。因为锌离子可在镁表面电沉积形成锌单质,与其周围的镁形成电偶腐蚀;同时锌离子的存在对镁表面附近的溶液具有缓冲作用,消耗镁腐蚀产生的氢氧根,使得pH值较长时间保持在7~8之间,促进氢氧化镁/氧化镁表面膜的溶解。所以这一机理可以很好地解释图 9中的结果。当较弱阳极极化下,锌离子较容易电沉积为锌单质(电沉积电位在-0.78 V附近),与镁形成电偶腐蚀,加速了析氢反应。强阳极极化时,锌离子就很难发生电沉积,微电偶的影响可以排除;同时锌离子的添加导致的溶液pH值较低使得表面膜产生较多孔隙,但是这很快被强阳极极化下产生的大量镁离子形成的产物膜堆积所抵消。

|
图 9 在氯化钠和氯化锌溶液中的析氢电流密度和阳极效率随极化电流密度的变化(保持析氢量相等) Fig. 9 Changes of the hydrogen evolution density and anode efficiency with the polarization current density in the NaCl and ZnCl2 solutions (keeping the amount of hydrogen evolution equal) |
图 10为在不同极化电流密度下氯化钠和氯化锌溶液中的阳极效率随极化时间的变化图。由图可知,随着时间的延长,阳极效率缓慢下降,最后保持平稳。在氯化锌溶液中,较大的阳极极化条件下,阳极效率的变化趋势与在氯化钠溶液中相似,但是较小的阳极极化条件下,氯化锌溶液中纯镁的阳极效率下降幅度较大,且持续时间较长。除了锌离子的沉积和pH值保持在较低水平共同作用外,实验误差也是原因之一。因为时间较短时,析氢量有限,实验误差偏高。阳极极化电流密度越大,析氢速率越快,测量误差越小,阳极效率也就越早进入平稳值。

|
图 10 在氯化钠(a)和氯化锌(b)溶液中的阳极效率随极化时间的变化 Fig. 10 Anode efficiency change with the polarization time in the NaCl(a) and ZnCl2 (b) solutions |
当然有些离子可能会对负差数效应产生相反的影响。研究表明[15],溶液中的In3+能够导致镁的阳极效率提高。目前其中的理论机制有待进一步研究。
3 分析与讨论金属镁作为牺牲阳极材料或空气电池的电极材料,其阳极效率的大小依赖于镁的阳极溶解反应,即阳极析氢或“负差数效应”。部分膜单价镁离子理论[16-17]认为,镁的表面存在一层不完整的多孔氧化膜,在膜破裂处或者膜的孔隙中,镁的阳极溶解过程分为化学过程和电化学过程。首先1 mol镁原子失去一个电子变为1 mol单价镁离子。然后k mol单价镁离子经过电化学反应转变为2价镁离子;剩余的(1-k) mol单价镁离子与水反应生成2价镁离子和氢氧根,并伴随着氢气产生,该析氢现象被称为阳极析氢。上述过程可表示为膜破裂或孔隙处的如下3个反应:
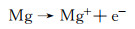
|
(2) |
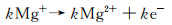
|
(3) |

|
(4) |
其中0≤k≤1。k是溶解的镁原子中通过电化学反应氧化成2价镁离子的百分数。实际镁在阳极溶解过程中,除了阳极析氢,还在裸露的基底处有阴极析氢,特别是当阳极极化弱时。阴极析氢反应可表示为:

|
(5) |
根据部分膜单价镁离子理论,镁的阳极效率η可定义为阳极极化电流(Ip)与按正常Faraday换算得到的总的溶解电流(IMg)的比。不考虑阴极过程时,
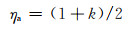
|
(6) |
电偶腐蚀放大效应是实际腐蚀的溶解量与电偶电流对应的溶解量的比。理论上,阳极效率的倒数是阳极电偶腐蚀的放大倍数(Ma):
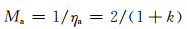
|
(7) |
因为通入(1+k) mol的电子,正常只能溶解(1+k)/2 mol当量的镁。但实际上却得到1当量的溶解量,溶解(腐蚀)被放大了[2/(1+k)]倍。
而当考虑阴极析氢时,实验所得阳极效率η则为:

|
(8) |
或

|
(9) |
式中:Vc为溶解1 mol镁原子时阴极析氢的体积;Vm是气体的摩尔体积(22.4 L/mol)。自腐蚀状态下,实验阳极效率最小,其值为0。实验电偶腐蚀放大倍数M则为:
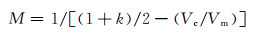
|
(10) |
由于电偶腐蚀放大作用与阳极效率有简单的倒数关系,后边的讨论重点将放在阳极效率上[16, 18]。可以看出,k值和Vc是决定效率或放大倍数的两个关键参数。其中k值与阳极析氢紧密相关,k值越大,阳极析氢的比例就越小,所以k值主要取决于极化电位、材料因素和表面膜。由于单价镁离子只能在膜破裂处裸露的镁基底上产生,穿过膜覆盖处溶解出来的都是2价镁,所以表面膜越完整、孔隙越少,k值就越大。Vc主要取决于阴极析氢,除了膜破裂处裸露的镁基底可以发生大量的阴极析氢,极薄的表面膜上也有可能发生阴极析氢,另外镁合金的杂质相和其他相上都可能发生阴极析氢。所以膜完整性对Vc的影响小于对k值的影响。另外阴极析氢所需的氢质子来自于溶液,而阳极析氢所需的水在溶液中大量存在,所以溶液成分对阴极析氢Vc的影响远大于阳极析氢。
3.2 阴阳极析氢、合金元素及第二相的影响图 2显示在盐酸溶液中阳极镁的析氢电流先减少后增大,分别由阴极析氢反应和阳极析氢反应主导,对应于纯镁的阳极效率先增大后稳定的过程。根据式(8),如果此时k值相对恒定,Vc决定于阳极效率的大小。随着极化电流的增大,阴极析氢电流降低,阳极效率增大;当测量电位趋近0时,阳极效率趋近ηa。而图 5中显示在腐蚀性较弱的氯化钠溶液中,纯镁的阴极析氢反应较弱,因此Vc影响可以忽略,阳极效率上升阶段也就不存在了。最终纯镁的阳极效率都稳定在一个理论值,该值大小取决于阳极析氢或k值,即负差数效应。同时随着腐蚀产物膜的产生,导致k值增大,进而引起阳极效率的略微下降。
纯镁本身的析氢过电位和对杂质的容忍度都较高,析氢较为困难。而镁合金表面存在微电偶,特别是第二相上的阴极析氢远比镁基体上容易[14],较多的阴极析氢使得镁合金的阳极效率较低。因此图 7中AZ91镁合金阳极效率上升阶段较纯镁要低。图 6展示了强阳极极化条件下局部微区的阴极析氢很难存在,因此镁阳极负差数效应与阴极微区(如杂质相、第二相)关系较小。宏观电偶效应(即阳极极化)较强时,镁合金的微电偶对阳极效率的影响不大。根据式(8),强阳极极化时,阴极析氢Vc趋于0,镁的阳极效率主要决定于k值。镁合金中的合金元素,可能直接影响式(2),(3)以及表面膜的完整性,进而影响镁的阳极效率。所有析氢过电位较镁基体低的其他相的存在,都会降低镁合金的阳极效率(图 7)。而固溶在镁基体相的合金元素对负差数效应和阳极效率也有重要的影响。如果这些元素可以促进具有致密性、保护性的表面膜的产生与存在,则可以提高k值。比如铝在第二相中提高了阴极析氢,但是固溶于镁基体中,则可以抑制阳极析氢。由于阳极效率同时决定于阴阳极析氢,同一元素可能在不同条件下具有不同的作用。合金化对镁电极阳极效率的影响,仍有大量的未知领域,需要进一步探究。
由图 8可知,硫酸溶液中纯镁的阳极效率要高于盐酸溶液中,这与氯离子比硫酸根离子更能促进阳极析氢有关。阳极效率也可能与溶液中阴离子的传递系数有关,阴离子可能影响到镁转化成单价镁离子以及单价镁离子转变为2价镁离子的过程。另外,图 9显示了含锌离子的溶液中弱阳极极化纯镁的析氢电流明显大于不含锌离子,而阳极效率较低。这是由于溶解到溶液中的锌离子可以通过电沉积和降低溶液pH值,破坏了表面的完整性,相当于减少了k值、增大了Vc,从而降低了阳极效率。但是强极化条件下表面腐蚀产物的堆积又会抵消它们的影响,因此强阳极极化下锌离子对阳极效率的影响不大。
3.3 镁牺牲阳极和镁空气电池的阳极效率镁合金作为牺牲阳极材料和镁空气电池的负极材料都已被广泛研究。因此可利用上述机理,进一步分析文献中的镁合金阳极效率较低的原因。
根据式(8),阳极效率决定于k值和Vc,它们与合金成分和相组成有关。一般认为具有低析氢过电位的合金元素的添加,会显著降低镁牺牲阳极效率;而具有高析氢过电位的贵金属元素铅、锡、镉、锌对镁阳极效率影响不大[19]。根据式(8),这些元素主要影响了Vc。一些合金元素的添加可以促进氧化膜的形成、降低镁合金的自腐蚀,提高阳极效率。如Jafari等[20]的研究表明,Zr和Be元素的添加可以提高AZ61镁合金的阳极效率。这相当于这些元素提高了k值,降低了Vc。相结构的改变也能极大地影响镁合金的阳极效率。比如铝元素的加入,可以通过形成Mg17Al12沉淀相减少活性区来提高镁阳极的效率[21]。有些元素的添加或者加工方式的改变还可以细化晶粒、调节第二相的分布,从而使得镁合金均匀腐蚀,避免了局部小颗粒的腐蚀脱落,以进一步提高阳极效率[3]。如Deng等[22]研究了空冷和水冷制备的Mg-0.5Ca合金的阳极性能,发现水冷的镁合金腐蚀更加均匀,具有更高的阳极利用率。这些加工方式的变化都直接或间接地优化了k值和Vc。
在镁空气电池的阳极效率的研究中,有研究表明[3, 23-24]阳极效率随着电流密度的增大逐渐增大并趋于平稳,这与式(8)预测的相一致。图 11为压力铸造和热轧AZ91D镁合金的阳极效率随极化电流密度变化图,由图可知,不同成型方式的AZ91D镁合金作为镁空气电池的阳极,它们的效率都随放电电流的增大而提高,又最终趋于一致;所不同的是低极化电流时,压铸态镁的阳极效率比热轧的高[3],因为热轧镁合金中含有更为分散的第二相颗粒,导致微电偶效应强烈。如2.2节所分析,微电偶效应随着极化电流的增加而逐渐被抑制,最终两个电极的阳极效率都达到一个稳定值,完全如式(8)所描述,也与镁合金自腐蚀越低则阳极析氢速率越低[25]的一般认知相符。当Mg-Ca-In镁合金作为镁空气电池的负极时,其阳极效率较Mg-Ca镁合金从62.7%提高到80.2%[25]。另外,In3+的添加也导致最终阳极效率的提高[15]。这些结果可能都是因为抑制了阳极析氢,具体机理尚需要进一步探讨。

(1) 镁的阳极效率受阳极析氢(负差数效应)控制,导致强极化条件下阳极效率无法达到百分之百,只能稳定在某一百分数。阴极析氢在弱极化条件下影响了阳极效率的大小。随着阳极化增强,阴极析氢对阳极效率的影响减弱,阳极析氢的影响增强。
(2) 随着阳极极化电流密度的增大,微电偶腐蚀的作用减弱,镁合金表面也趋于均匀化。强极化时,微电偶并不是导致镁阳极效率低的原因。但弱极化时或自腐蚀条件下,微电偶对镁阳极效率具有显著影响。
(3) 溶液中离子会影响到表面膜的保护性、pH值以及镁的阳极溶解的中间过程,从而改变镁的阳极效率。
(4) 在镁牺牲阳极保护系统和镁空气电池中,当镁合金阳极处于强极化工作状态时,阳极析氢限制了镁电极阳极效率的最高值。而当镁阳极处于非工作或存储状态时,阴极析氢可以进一步降低其阳极效率。
| [1] |
YAN L, SONG G L, ZHENG D. Magnesium alloy anode as a smart corrosivity detector and intelligent sacrificial anode protector for reinforced concrete[J]. Corrosion Science, 2019, 155: 13-28. DOI:10.1016/j.corsci.2019.04.027 |
| [2] |
WANG F, XU J, XU Y, et al. A comparative investigation on cathodic protections of three sacrificial anodes on chloride-contaminated reinforced concrete[J]. Construction and Building Materials, 2020, 246: 118476. DOI:10.1016/j.conbuildmat.2020.118476 |
| [3] |
XIAO B, SONG G L, ZHENG D, et al. A corrosion resistant die-cast Mg-9Al-1Zn anode with superior discharge performance for Mg-air battery[J]. Materials & Design, 2020, 194: 108931. |
| [4] |
VAGHEFINAZARI B, HÖ CHE D, LAMAKA S V, et al. Tailoring the Mg-air primary battery performance using strong complexing agents as electrolyte additives[J]. Journal of Power Sources, 2020, 453: 227880. DOI:10.1016/j.jpowsour.2020.227880 |
| [5] |
SONG G L, ATRENS A. Corrosion mechanisms of magnesium alloys[J]. Advanced Engineering Materials, 1999, 1(1): 11-33. DOI:10.1002/(SICI)1527-2648(199909)1:1<11::AID-ADEM11>3.0.CO;2-N |
| [6] |
SONG G L, ATRENS A, STJOHN D, et al. The electrochemical corrosion of pure magnesium in 1 N NaCl[J]. Corrosion Science, 1997, 39(5): 855-875. DOI:10.1016/S0010-938X(96)00172-2 |
| [7] |
HUANG J F, SONG G L, ZHU Y X, et al. The anodically polarized Mg Surface products and accelerated hydrogen evolution[J]. Journal of Magnesium and Alloys. DOI:10.1016/j.jma.2016.05.008 |
| [8] |
SONG G L, JOHANNESSON B, HAPUGODA S, et al. Galvanic corrosion of magnesium alloy AZ91D in contact with an aluminium alloy, steel and zinc[J]. Corrosion Science, 2004, 46(4): 955-977. DOI:10.1016/S0010-938X(03)00190-2 |
| [9] |
SONG G L, ATRENS A, St JOHN D. An hydrogen evolution method for the estimation of the corrosion rate of magnesium alloys[C]//TMS.Magnesium Technology.United States: John Wiley & Sons, 2001: 255-262.
|
| [10] |
SONG G L, St JOHN D. The effect of zirconium grain refinement on the corrosion behaviour of magnesium-rare earth alloy MEZ[J]. Journal of Light Metals, 2002, 2(1): 1-16. DOI:10.1016/S1471-5317(02)00008-1 |
| [11] |
SONG G L, ATRENS A, St JOHN D, et al. The anodic dissolution of magnesium in chloride and sulphate solutions[J]. Corrosion Science, 1997, 39(10/11): 1981-2004. |
| [12] |
SONG G L, ATRENS A, WU X, et al. Corrosion behaviour of AZ21, AZ501 and AZ91 in sodium chloride[J]. Corrosion Science, 1998, 40(10): 1769-1791. DOI:10.1016/S0010-938X(98)00078-X |
| [13] |
CHU P, LE MIRE E, MARQUIS E A. Microstructure of localized corrosion front on Mg alloys and the relationship with hydrogen evolution[J]. Corrosion Science, 2017, 128: 253-264. DOI:10.1016/j.corsci.2017.09.022 |
| [14] |
HUANG J, SONG G, WANG Z, et al. The Zn2+ destabilized surface film and accelerated corrosion of magnesium[J]. Journal of the Electrochemical Society, 2020, 167(16): 161508. DOI:10.1149/1945-7111/abd002 |
| [15] |
GORE P, FAJARDO S, BIRBILIS N, et al. Anodic activation of Mg in the presence of In3+ ions in dilute sodium chloride solution[J]. Electrochimica Acta, 2019, 293: 199-210. DOI:10.1016/j.electacta.2018.09.155 |
| [16] |
HUANG J, SONG G L, ATRENS A, et al. What activates the Mg surface-a comparison of Mg dissolution mechanisms[J]. Journal of Materials Science & Technology, 2020, 57: 204-220. |
| [17] |
CAIN T W, GLOVER C F, LAIRD J S, et al. Insight into the effect of Mg(OH)2 films vs noble element enrichment on the global and local cathodic activation of corroding Mg[J]. Corrosion, 2020, 77(2): 115-133. |
| [18] |
HUANG J F, SONG G L, ZHU Y X, et al. The anodically polarized Mg surface products and accelerated hydrogen evolution[J]. Jounal of Magesium and Alloys. DOI:10.1016/j.jma.2021.05.008 |
| [19] |
MORGAN J. Cathodic protection[M]. Houston: NACE International, 1987.
|
| [20] |
JAFARI H, MOHAMMAD HASSANIZADEH B. Influence of Zr and Be on microstructure and electrochemical behavior of AZ63 anode[J]. Materials and Corrosion, 2018, 70(4): 633-641. |
| [21] |
MA J, WANG G, LI Y, et al. Electrochemical performance of Mg-air batteries based on AZ series magnesium alloys[J]. Ionics, 2018, 25(5): 2201-2209. DOI:10.1007/s11581-018-2705-1?utm_source=xmol |
| [22] |
DENG M, HÖCHE D, LAMAKA S V, et al. Revealing the impact of second phase morphology on discharge properties of binary Mg-Ca anodes for primary Mg-air batteries[J]. Corrosion Science, 2019, 153: 225-235. DOI:10.1016/j.corsci.2019.03.050 |
| [23] |
CHEN X, LIAO Q, LE Q, et al. The influence of samarium (Sm) on the discharge and electrochemical behaviors of the magnesium alloy AZ80 as an anode for the Mg-air battery[J]. Electrochimica Acta, 2020, 348: 136315. |
| [24] |
CHEN X, WANG H, ZOU Q, et al. The influence of heat treatment on discharge and electrochemical properties of Mg-Gd-Zn magnesium anode with long period stacking ordered structure for Mg-air battery[J]. Electrochimica Acta, 2021, 367: 137518. DOI:10.1016/j.electacta.2020.137518 |
| [25] |
DENG M, WANG L, HÖCHE D, et al. Ca/In micro alloying as a novel strategy to simultaneously enhance power and energy density of primary Mg-air batteries from anode aspect[J]. Journal of Power Sources, 2020, 472: 228528. DOI:10.1016/j.jpowsour.2020.228528 |