 2021, Vol. 49
2021, Vol. 49文章信息
- 杨夕馨, 常增花, 邵泽超, 吴帅锦, 王仁念, 王建涛, 卢世刚
- YANG Xi-xin, CHANG Zeng-hua, SHAO Ze-chao, WU Shuai-jin, WANG Ren-nian, WANG Jian-tao, LU Shi-gang
- 富锂锰基正极材料在不同温度下的极化行为
- Polarization behavior of lithium-rich manganese-based cathode materials at different temperatures
- 材料工程, 2021, 49(9): 69-78
- Journal of Materials Engineering, 2021, 49(9): 69-78.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2020.000376
-
文章历史
- 收稿日期: 2020-04-26
- 修订日期: 2020-10-15




2. 国联汽车动力电池研究院有限责任公司, 北京 100088;
3. 北京有色金属研究总院, 北京 100088
2. China Automotive Battery Research Institute Co., Ltd., Beijing 100088, China;
3. General Research Institute for Nonferrous Metals, Beijing 100088, China
锂离子电池以其具有高比能量及功率密度、长循环寿命、环境友好等特点在消费类电子产品、电动汽车和储能等领域得到了广泛的应用[1-2]。为了满足日益增长的应用需求,开发新型高比能量正极材料是提升锂离子电池性能的关键环节。层状富锂锰基正极材料因其具有高的放电比容量成为最具有前景的正极材料之一[3-4]。目前,对富锂锰基正极材料在常温下的研究已经取得一定进展,表面包覆[5-6]、体相掺杂[7-8]、材料形貌控制[9-10]、电极结构设计[11]等方法使材料性能有了很大的提升。然而,富锂锰基材料的电化学性能在不同温度下会表现出巨大的差异,Yu等[12]的研究表明,0.3Li2MnO3·0.7LiMn1/3Ni1/3Co1/3O2在45 ℃下放电容量为238 mAh/g,而-10 ℃时仅为114.5 mAh/g。Vivekanantha等[13]发现Li1.2Mn0.54Ni0.13Co0.13O2在5, 25 ℃和55 ℃下放电容量差异很大,分别为101, 190 mAh/g和274 mAh/g。材料性能随温度变化的差异会严重限制材料在温度多变的实际环境中的应用。因此,研究温度对富锂锰基材料电化学性能的影响规律及其影响因素,可为富锂锰基材料环境适应性的改善提供理论基础。有研究者认为在充放电过程中的电荷转移过程是不同温度下富锂锰基材料性能存在差异的主要影响步骤[14-15],因此通过在富锂锰基材料中掺杂Co离子使反应活化能降低,从而使材料性能提升[13]。也有研究者认为不同环境温度下材料的性能主要依赖于锂离子在材料中的扩散,因此基于锂离子扩散系数与层间距的关系优化了材料的温度效应[16-17]。虽然这些工作在一定程度上降低了材料对温度的敏感性,提升了材料的性能,但是研究者对引起材料在不同温度下电化学性能差异的影响因素尚未有统一和深入的认识,而这对于推动富锂锰基材料在实际中的应用是非常必要的。因此,本工作将通过恒电流间歇滴定测试(GITT)和电化学阻抗谱(EIS)等电化学测试方法表征不同温度下富锂锰基材料本征的极化特性并且系统地研究材料性能依赖于温度的影响因素,为进一步改善富锂锰基材料的电化学性能提供理论依据。
1 实验材料与方法 1.1 电池组装将本课题组自行研制的正极活性材料Li1.18Mn0.5Ni0.26Co0.06O2、导电剂(导电石墨KS-6、导电炭黑SP、碳纳米管CNTs的比例为1∶ 2∶1)和黏结剂聚偏氟乙烯(PVDF)按质量比94∶4∶2充分混合,并加入适量溶剂N-甲基吡咯烷酮(NMP)制成浆料。待正极浆料混合均匀后涂布在铝箔上,将涂覆好的极片置于80 ℃的烘箱中干燥6 h,使用冲片机制得直径为14 mm的正极片,将正极片辊压至一定的压实密度(2.6 g/cm3)后,置于100 ℃的真空烘箱中干燥12 h,得到电池正极片。以所得极片为正极,金属锂为负极,Celgard2400聚丙烯薄膜为隔膜,加入适量电解液后,在高纯氩气氛围的手套箱(氧气含量以及水含量均小于0.1×10-6)中,组装得到CR2032型扣式半电池。
1.2 电化学性能测试使用生化培养箱(BPMJ-500F)控制环境温度,使用LAND电池测试系统(CT2001A),以0.1 C的电流密度(1 C=227 mAh/g),在2.0~4.8 V的电压范围内对扣式半电池进行不同温度条件下的充放电测试和恒电流间歇滴定(GITT)测试,GITT测试恒电流脉冲时间为10 min,静置时间为40 min。使用电化学工作站(MetrohmAutolab B.V.)对扣式半电池进行不同温度下的电化学阻抗谱(EIS)测试,测试前在设定的温度下保温2 h以确保电池内部达到平衡,电压振幅为5 mV,测试频率范围为106~10-2 Hz。需要指出的是,本工作通过GITT测试得到的极化值是电极的总极化值,包括欧姆降、电化学极化、浓差极化等,如图 1所示,以0.1 C恒流充/放电10 min后得到闭路电压(CCV),静置40 min后得到可近似等于平衡电压的开路电压(OCV),极化值定义为各电压暂态下CCV与OCV的差值。

|
图 1 GITT曲线及极化值示意图 Fig. 1 GITT curve and polarization value diagram |
选取测试样品在指定温度环境下循环至容量趋于稳定的第5周充放电过程进行电化学极化行为研究。图 2(a)为富锂锰基材料在不同温度下的充放电曲线,在45, 35, 25, 15 ℃和5 ℃下的充电比容量分别为266.6, 240.0, 216.7, 177.6 mAh/g和165.7 mAh/g,放电比容量分别为257.5, 231.3, 209.3, 172.6 mAh/g和162.1 mAh/g,显然温度对材料的充放电比容量有很大影响。为了进一步了解温度对电化学反应过程的影响,对充放电曲线进行微分计算,得到不同温度下的容量微分曲线,并根据文献报道[18-21]将各峰对应的电化学反应区间进行划分,如图 2(b)所示。充电容量微分曲线中,Ⅰ峰主要为氧、锰离子氧化反应(锰离子反应是由材料初始充电高于4.5 V时Li2MnO3组分活化,发生氧损失所造成),Ⅱ峰主要为镍、钴离子反应,且伴随有部分氧参与电荷补偿反应,Ⅲ峰对应于氧的电荷补偿反应[18-19];在放电过程中,Ⅳ,Ⅵ峰对应氧的还原反应,Ⅴ峰对应镍、钴离子还原反应,少量锰离子在更低位下参与电化学反应[20-21]。随着温度的变化,Ⅰ,Ⅲ和Ⅵ峰的峰强度呈现显著变化,温度越低,峰强度越低,且Ⅰ峰的峰位向高电位偏移,而Ⅱ, Ⅴ峰呈现出的差异相对较小。这意味着在充放电过程中镍/钴离子的电化学反应受温度影响较小,而氧/锰离子的电化学反应对温度变化非常敏感,较高温度45 ℃下反应能有利地进行,而在较低温度5 ℃下反应严重受阻。

|
图 2 不同温度下的充放电曲线 (a)电压-容量曲线; (b)容量微分曲线 Fig. 2 Charge and discharge curves at different temperatures (a)voltage-capacity curve; (b)capacity differential curve |
区分不同温度充放电时在不同电压区间内各反应贡献的容量可以更清楚地观察到氧/锰离子反应受温度影响更大。如图 3所示,在对应于镍/钴离子反应的充电中压区间3.61~4.10 V,以及放电中压区间4.00~3.53 V内,随温度的改变,比容量的变化程度很小,充电比容量维持在100~120 mAh/g,放电比容量维持在80~90 mAh/g。然而,在对应于氧离子参与反应的充放电高压区间内,比容量受温度的影响较大;在充电(开始充电~3.61 V)和放电(3.53 V~放电结束)的低电压区间内,锰离子参与反应使比容量受温度的限制更为明显,较低温度5 ℃下的充、放电比容量3.2, 44.7 mAh/g仅为在室温25 ℃下充、放电比容量23.4, 71.2 mAh/g的13.7%及62.8%,而在较高温度45 ℃下充、放电比容量53.5, 124.4 mAh/g可以达到室温25 ℃下的2.3倍和1.8倍。

|
图 3 不同温度下充电(a)及放电(b)过程中各电压区间内反应贡献的比容量对比 Fig. 3 Specific capacity comparison of reaction contribution in each voltage range during charging (a) and discharging (b) at different temperatures |
目前普遍认为不同温度下锂离子电池的电化学性能具有较大差异主要与动力学因素有关[22],因此本工作采用GITT表征了不同温度下富锂锰基材料的极化特性,如图 4所示。在充电过程中,各温度下的极化值曲线随着OCV的增加呈现出“U”形变化趋势;在放电过程中各极化值在较高电压尤其是中低电压区间较大,在放电结束时快速增大。与图 2(b)对比可以发现,极化值较大的电压区间对应的是氧/锰离子的氧化还原反应,这意味着与镍/钴离子较快的电化学反应动力学相对比,氧/锰离子的反应动力学缓慢。此外,从图 4可观察到在充放电过程中,随着温度从45 ℃降低至5 ℃,极化值逐渐增加,但各区间对应的极化值增加量并不相同。在充电过程中(图 4(a)),在3.8~4.2 V电压范围内极化值增加量较小,约为0.02~0.05 V,而在低于3.7 V的电压区间和高于4.3 V的电压区间内极化值增加明显,分别约为0.05~0.08 V和0.07~0.22 V。在放电过程中(图 4(b)),同样呈现出在高电压和低电压区间内极化值的增加量显著高于其他电压区间的特征,在电压高于4.2 V时约为0.05~0.09 V,在4.2~3.8 V电压范围内约为0.03~0.05 V,在低于3.8 V时约为0.05~1.10 V。

|
图 4 不同温度下充电(a)及放电(b)过程的极化值变化曲线 Fig. 4 Polarization value change curve during charging (a) and discharging (b) at different temperatures |
上述极化规律与材料充放电容量随温度的变化规律一致,从45 ℃至5 ℃,极化值逐渐增加,材料比容量逐渐降低,且在极化值变化较小的电压区间内,材料容量受温度的影响较小,而在极化值变化明显的电压区间,材料容量受到环境温度的限制。由此可知,不同温度下富锂锰基材料的性能与电化学反应动力学,尤其是高电压和低电压区间内的氧/锰离子反应动力学特征密切相关。
2.3 影响富锂锰基材料极化-温度依赖性的因素电化学反应包括锂离子液相传输、界面锂离子传输、电荷转移以及锂离子固相扩散过程。为了进一步追溯影响富锂锰基材料动力学特性与温度依赖关系的电极过程,本工作借助交流阻抗分析获得不同过程的阻抗,进而利用阿仑尼乌斯方程计算得到不同过程的活化能,以确定不同电化学反应对应的电极过程对温度的依赖性。
测定了不同OCV的扣式半电池在不同温度条件下的电化学阻抗,并进行了拟合分析,如图 5和图 6所示。不同OCV下,交流阻抗谱随温度的变化规律相似。较高温度下交流阻抗谱主要由中高频区的两个半圆和低频区的一条斜线构成。通常认为高频区的半圆与锂离子在固相/电解质界面膜(SEI膜)中的传输有关,中频区的半圆与电荷转移过程有关,低频区的斜线与锂离子固相扩散有关。随着温度的降低,相同频率区间阻抗谱的变化主要表现出以下3个特征:(1)整个曲线向右移动,说明Li+在电解液中的传输阻抗增加(对应于电解液电导率的降低);(2)中高频区的圆弧半径明显增大,这表明界面锂离子传输阻抗和电荷转移阻抗显著增加;(3)低频区与Li+在电极中扩散相关的斜线逐渐变短,到15 ℃时基本消失。

|
图 5 充电和放电过程中不同OCV的扣式电池在不同温度下的电化学阻抗谱 (a)charge 3.41 V; (b)charge 3.75 V; (c)charge 4.45 V; (d)discharge 4.33 V; (e)discharge 3.75 V; (f)discharge 3.39 V Fig. 5 Electrochemical impedance spectroscopy of coin cells of different OCV at different temperatures during charging and discharging process (a)charge 3.41 V; (b)charge 3.75 V; (c)charge 4.45 V; (d)discharge 4.33 V; (e)discharge 3.75 V; (f)discharge 3.39 V |

|
图 6 充电和放电过程中在不同OCV下阻抗值随温度的变化 (a)charge OCV=3.41 V; (b)charge OCV=3.75 V; (c)charge OCV=4.43 V; (d)discharge OCV=4.33 V; (e)discharge OCV=3.75 V; (f)discharge OCV=3.37 V Fig. 6 Changes of impedance value with temperature at different OCV during charging and discharging process (a)charge OCV=3.41 V; (b)charge OCV=3.75 V; (c)charge OCV=4.43 V; (d)discharge OCV=4.33 V; (e)discharge OCV=3.75 V; (f)discharge OCV=3.37 V |
对阻抗测试结果按照图 6(a)所示的等效电路进行拟合分析[23],结果如图 6所示,其中Rb为内阻,RSEI为SEI膜阻抗,Rct为电荷转移阻抗。由图 6可知,在各OCV下Rb随温度变化较小,即与RSEI和Rct相比,温度对Rb的影响可以忽略。而RSEI及Rct均随温度的降低呈现出增加的趋势,会造成在较低温度下反应时呈现出较大的极化,与图 4结果一致。此外,还发现在不同OCV下随着温度的降低RSEI及Rct的增加量并不相同,从45 ℃到5 ℃,在充电过程OCV为3.41, 3.75, 4.45 V时和放电过程OCV为4.33, 3.75, 3.39 V时,RSEI的增加量分别为983, 252, 128 Ω和228, 329, 613 Ω,Rct的增加量分别为3890, 290, 2792 Ω和1793, 503, 2185 Ω。可以发现,RSEI在充放电的低电压区间比在高电压区间随温度变化程度更大;Rct在高电压及低电压区间改变量较大,而在中间电压区间改变量较小。由于较低温度下阻抗谱中与固相扩散相关的斜线消失,无法得到富锂锰基材料在不同OCV下固相扩散随温度的变化规律,因此基于GITT数据通过式(1)计算得到在不同温度下充放电时的锂离子扩散系数[24],如图 7所示。在测试温度范围内,5 ℃下的锂离子扩散系数在充放电过程中均为最小,随着温度的升高,锂离子扩散系数逐渐增大。此外,还发现随着温度的降低,锂离子扩散系数的降低程度在各OCV区间内也表现出差异,在充电(3.0~4.0 V)和放电(4.6~4.0 V)的初始阶段,锂离子扩散系数降低程度较小,而在充电(4.2~4.6 V)和放电(3.5~3.0 V)后期,锂离子扩散系数大幅降低。

|
(1) |

|
图 7 充电(a)及放电(b)过程中不同温度下的锂离子扩散系数变化曲线 Fig. 7 Curves of lithium ion diffusion coefficient at different temperatures during charging (a) and discharging (b) process |
式中:m和M分别是正极活性材料的质量和相对分子质量; Vm是正极材料的摩尔体积; S是电极和电解液之间的总接触面积;τ是弛豫时间。ΔEτ和ΔEs分别表示单次电流脉冲后电压的变化以及相邻两次开路电压的变化,如图 1所示。计算锂离子扩散系数DLi+时假设Vm随着荷电状态(SOC)的变化保持不变,由于电极完全浸在电解液中,电解液很容易渗透到导电剂和黏结剂中,因此比表面积可以很好地近似于活性材料与电解液的总接触面积,常用于计算锂离子扩散系数[24],该富锂正极材料的比表面积为1.316 m2/g。
由各电极过程的活化能可以更清晰地了解其与温度的依赖关系,活化能可由式(2), (3)和式(4)计算[25]:
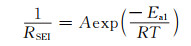
|
(2) |
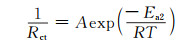
|
(3) |
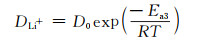
|
(4) |
式中: A, Ea, R, T分别表示频率因子,活化能,气体常数,绝对温度;D0为本征扩散系数。
图 8为充放电过程中不同OCV下电荷转移过程阻抗、界面锂离子传输阻抗以及锂离子固相扩散系数(1/RSEI,1/Rct和DLi+)与温度的阿伦尼乌斯关系图。随着充电电压升高,锂离子在SEI膜中扩散过程的表观活化能为60.45, 56.60, 54.12 kJ/mol,电荷转移过程的表观活化能为71.68, 67.69, 77.63 kJ/mol;随着放电过程进行,上述两个过程的表观活化能分别为57.79, 61.89, 64.32 kJ/mol和75.83, 73.78, 79.99 kJ/mol。另外,在充放电过程中各选取了相应OCV下的锂离子扩散系数计算表观活化能。充电过程中,OCV为3.61, 3.93 V和4.44 V时锂离子固相扩散表观活化能分别为7.97, 12.95 kJ/mol和50.35 kJ/mol;放电过程中,OCV为4.31, 3.76 V和3.34 V时锂离子固相扩散表观活化能分别为18.01, 24.95 kJ/mol和54.46 kJ/mol。

|
图 8 充电(1)和放电(2)过程中不同OCV下不同电极过程的表观活化能 (a)电荷转移过程表观活化能;(b)界面锂离子传输表观活化能;(c)锂离子固相扩散表观活化能 Fig. 8 Apparent activation energy of different electrode processes at different OCV during charge (1) and discharge (2) process (a)apparent activation energy of charge transfer; (b)apparent activation energy of interfacial lithium ion transfer; (c)apparent activation energy of lithium ion solid phase diffusion |
由不同OCV下各电极过程的表观活化能规律可知,电荷转移过程对应的表观活化能最高,其次是界面锂离子传输对应的表观活化能,而锂离子固相扩散对应的表观活化能仅在充放电末期具有较高的数值,其他电压区间对应的表观活化能远低于前两者。这说明富锂锰基材料的充放电性能与温度的依赖关系主要取决于电荷转移过程,其次是界面锂离子传输过程。此外,结合图 2中Ⅰ, Ⅲ和Ⅵ峰的峰强度与温度具有强依赖关系的特点与图 9中与Ⅰ, Ⅲ和Ⅵ峰对应OCV下的表观活化能不难发现,Ⅵ峰的峰强度随温度变化最显著,原因在于界面锂离子传输、电荷转移以及锂离子固相扩散均具有较高的表观活化能。Ⅰ峰的峰强度与温度的依赖关系主要受前两个过程的影响,Ⅲ峰的峰强度与温度的依赖关系与三个过程均具有较强的相关性,与Ⅰ峰相比,只是界面锂离子传输对应的表观活化能有所降低。上述三个电极过程的表观活化能随OCV的变化规律涉及充放电过程中材料结构的演化。

|
图 9 充电(a)及放电(b)过程中各电极过程的表观活化能 Fig. 9 Apparent activation energy of each electrode process during charging (a) and discharging (b) process |
与高电压区间相比,低电压区间内锂离子在SEI膜中的扩散过程呈现出更高的能量势垒,这可能与在充放电过程中SEI膜的化学组成变化有关[26]。低电位下充电时发生氧(可能还包含锰)的氧化反应,放电时氧(可能还包含锰)发生还原反应,也就是说氧(可能还包含锰)的电荷补偿反应对应的电压区间表面阻抗RSEI较高。导致这一结果的原因可能是由于氧/锰和镍/钴的电荷补偿对应于材料中不同结构构型,材料表面也具有不同的组成和结构[27],造成所形成的固/液界面也有所区别[28],在能够发生锰电荷补偿反应的结构构型中固/液界面内锂离子扩散阻抗较高,从而使得充电过程高电压下呈现出较小的表观活化能[29]。
与此同时,电荷转移反应的表观活化能在充放电的高电压与低电压下较高,而在中压3.75 V时较低,这种差异与不同OCV下发生了不同的电化学反应且不同电化学反应的本征反应动力学性能不同有关[30];对比容量微分图(图 2(b))可知,具有较低表观活化能的充放电中间电压对应的是快动力学的镍/钴离子反应区间,而具有较高表观活化能的高电压及低电压对应的是缓慢动力学的氧/锰离子电荷补偿反应。
锂离子固相扩散表观活化能随OCV的变化规律与充放电过程中材料结构的变化以及锂离子浓度的改变有关[31-32]。在充电初期,对应氧/锰离子的氧化反应,锂离子从较为复杂的类尖晶石结构中脱出[33],扩散动力学相对较差,但是充电初始阶段锂离子从高锂离子浓度的富锂材料中脱出容易进行,表观活化能较低;在充电的中间电压区间,对应镍/钴离子的氧化,锂离子从层状结构中脱出过程较为容易[30],但富锂材料中锂离子的浓度有所降低使该阶段与充电初期相比表观活化能略有增大;在充电末期,对应氧的电荷补偿反应[30],剩余的锂离子从过渡金属层中脱出,过渡金属的存在阻碍锂离子的迁移过程,且经历了充电前中期材料结构的变化,此时材料存在多种相结构[34],锂离子的脱出环境较为复杂,除此之外,充电末期富锂材料中剩余的锂离子较少,锂离子的脱出过程不易进行,造成充电末期锂离子扩散表观活化能大幅增加。在放电过程中,高压区间内参与还原反应的是性能相对较好的可逆氧[35],且放电初期锂离子嵌入低锂离子浓度的富锂锰基材料中较容易发生,此时的表观活化能在整个放电过程中相对较小;中电压区内由镍/钴离子进行还原反应,锂离子嵌入层状结构较容易[30],但是,随着反应进行,富锂锰基材料中锂离子浓度增加,剩余的锂离子嵌入阻力增加,造成表观活化能与放电初期相比略有增加;低电压区对应的是氧/锰离子的反应,在这一阶段,锂离子嵌入到部分类尖晶石结构中,材料结构变化十分复杂[36],锂离子扩散受到较大阻力,且放电末期锂离子进一步嵌入锂空位较少的富锂锰基材料时动力学阻碍变大,造成这一阶段的表观活化能明显增大。
综上可知,锂离子在SEI膜中的传输过程、电荷转移过程以及锂离子的固相扩散过程对富锂锰基材料电化学性能与温度的依赖关系产生了不同程度的影响。其中,充放电过程中的电荷转移尤其是氧/锰离子反应的电荷转移动力学特性差是造成富锂锰基材料性能受温度限制的主要因素。其次,在低电压区间氧电荷补偿反应使材料结构发生复杂变化从而诱发表面膜成分变化,使锂离子在SEI膜中传输动力学变差,对温度变化更为敏感,导致低电压区间极化值随温度变化程度大。此外,在充放电末期氧/锰离子参与反应使材料结构发生复杂变化,导致锂离子扩散系数对温度变化更敏感,进而使充放电末期极化值随温度变化程度进一步增大。上述因素均导致氧/锰离子贡献的容量与温度具有较强的依赖关系,这是造成富锂锰基材料充放电容量随温度变化较大的主要原因。
3 结论(1) 随着温度的降低,富锂锰基材料充放电过程极化明显增大,尤其在充放电高电压及低电压区间内氧/锰离子反应的极化增加程度更大。
(2) 低温下较大的极化造成富锂锰基材料充放电容量显著降低,与45 ℃相比,在5 ℃下富锂锰基材料充放电容量分别降低100.9 mAh/g和95.4 mAh/g。这主要是因为氧/锰离子本征的动力学反应差使富锂锰基材料在高电压及低电压区间具有较高的电荷转移表观活化能。
(3) 此外,氧/锰离子反应使材料结构发生复杂变化,诱发SEI膜成分发生变化,使其在低电压下界面锂离子传输表观活化能增大,并且在该结构中锂离子固相扩散表观活化能显著升高。因此,建议未来的工作可聚焦于改善富锂锰基材料中氧、锰反应动力学来提高其环境适应性。
| [1] |
DUNN B, KAMATH H, TARASCON J M. Electrical energy storage for the grid: a battery of choices[J]. Science, 2011, 334(6058): 928-935. DOI:10.1126/science.1212741 |
| [2] |
YANG Z, ZHANG J, KINTNER-MEYER M C W, et al. Electrochemical energy storage for green grid[J]. Chemical Reviews, 2011, 111(5): 3577-3613. DOI:10.1021/cr100290v |
| [3] |
ZHAO T, JI R, MENG Y. The role of precipitant in the preparation of lithium-rich manganese-based cathode materials[J]. Chemical Physics Letters, 2019, 730: 354-360. DOI:10.1016/j.cplett.2019.06.034 |
| [4] |
DENG B, CHEN Y, WU P, et al. Lithium-rich layered oxide nanowires bearing porous structures and spinel domains as cathode materials for lithium-ion batteries[J]. Journal of Power Sources, 2019, 418: 122-129. DOI:10.1016/j.jpowsour.2019.02.036 |
| [5] |
HU S, LI Y, CHEN Y, et al. Insight of a phase compatible surface coating for long-durable Li-rich layered oxide cathode[J]. Advanced Energy Materials, 2019, 9(34): 1901795. DOI:10.1002/aenm.201901795 |
| [6] |
蔺佳明, 赵桃林, 王育华, 等. Li2ZrO3包覆锂离子电池正极材料Li[Li0.2Ni0.2Mn0.6]O2的制备及其电化学性能[J]. 材料工程, 2020, 48(3): 112-120. LIN J M, ZHAO T L, WANG Y H, et al. Fabrication and electrochemical performance of Li[Li0.2Ni0.2Mn0.6]O2 coated with Li2ZrO3 as cathode material for lithium-ion batteries[J]. Journal of Materials Engineering, 2020, 48(3): 112-120. |
| [7] |
WANG M J, YU F D, SUN G, et al. Co-regulating the surface and bulk structure of Li-rich layered oxides by a phosphor doping strategy for high-energy Li-ion batteries[J]. Journal of Materials Chemistry A, 2019, 7(14): 8302-8314. DOI:10.1039/C9TA00783K |
| [8] |
BAO L, YANG Z, CHEN L, et al. The effects of trace Yb doping on the electrochemical performance of Li-rich layered oxides[J]. ChemSusChem, 2019, 12(10): 2294-2301. DOI:10.1002/cssc.201900226 |
| [9] |
WU Z L, XIE H, LI Y, et al. Li1.2Ni0.25Mn0.55O2: a high-capacity cathode material with a homogeneous monoclinic Li2MnO3-like superstructure[J]. Journal of Alloys and Compounds, 2020, 827: 154202. DOI:10.1016/j.jallcom.2020.154202 |
| [10] |
JIANG X, WANG Z, ROONEY D, et al. A design strategy of large grain lithium-rich layered oxides for lithium-ion batteries cathode[J]. Electrochimica Acta, 2015, 160: 131-138. DOI:10.1016/j.electacta.2015.02.061 |
| [11] |
黄贤凯, 邵泽超, 常增花, 等. 导电炭黑对富锂锰基层状氧化物电极性能的影响[J]. 材料工程, 2019, 47(8): 13-21. HUANG X K, SHAO Z C, CHANG Z H, et al. Effect of conductive carbon black on electrochemical performance of Li- and Mn-rich layered oxide electrode[J]. Journal of Materials Engineering, 2019, 47(8): 13-21. |
| [12] |
YU C, WANG H, GUAN X, et al. Conductivity and electrochemical performance of cathode xLi2MnO3·(1-x) LiMn1/3Ni1/3Co1/3O2 (x=0.1, 0.2, 0.3, 0.4) at different temperatures[J]. Journal of Alloys and Compounds, 2013, 546: 239-245. DOI:10.1016/j.jallcom.2012.08.026 |
| [13] |
VIVEKANANTHA M, SENTHIL C, KESAVAN T, et al. Reactive template synthesis of Li1.2Mn0.54Ni0.13Co0.13O2 nanorod cathode for Li-ion batteries: influence of temperature over structural and electrochemical properties[J]. Electrochimica Acta, 2019, 317: 398-407. DOI:10.1016/j.electacta.2019.05.095 |
| [14] |
KOU J, CHEN L, SU Y, et al. The role of cobalt content in improving low temperature performance of layered lithium-rich cathode materials for Li-ion batteries[J]. ACS Applied Materials & Interfaces, 2015, 7(32): 17910-17918. |
| [15] |
YU H, WANG Y, ASAKURA D, et al. Electrochemical kinetics of the 0.5Li2MnO3·0.5LiMn0.42Ni0.42Co0.16O2 'composite' layered cathode material for lithium-ion batteries[J]. RSC Advances, 2012, 2(23): 8797-8807. DOI:10.1039/c2ra20772a |
| [16] |
YANG S, YAN B, WU J, et al. Temperature-dependent lithium-ion diffusion and activation energy of Li1.2Co0.13Ni0.13Mn0.54O2thin-film cathode at nanoscale by using electrochemical strain microscopy[J]. ACS Applied Materials & Interfaces, 2017, 9(16): 13999-14005. |
| [17] |
CUI S, WEI Y, LIU T, et al. Optimized temperature effect of Li-ion diffusion with layer distance in Li(NixMnyCoz)O2 cathode materials for high performance Li-ion battery[J]. Advanced Energy Materials, 2016, 6(4): 1501309. DOI:10.1002/aenm.201501309 |
| [18] |
YABUUCHI N, YOSHII K, MYUNG S T, et al. Detailed studies of a high-capacity electrode material for rechargeable batteries, Li2MnO3-LiCo1/3Ni1/3Mn1/3O2[J]. Journal of the American Chemical Society, 2011, 133(12): 4404-4419. DOI:10.1021/ja108588y |
| [19] |
THACKERAY M M, KANG S H, JOHNSON C S, et al. Comments on the structural complexity of lithium-rich Li1+xM1-xO2 electrodes (M=Mn, Ni, Co) for lithium batteries[J]. Electrochemistry Communications, 2006, 8(9): 1531-1538. DOI:10.1016/j.elecom.2006.06.030 |
| [20] |
SATHIYA M, ROUSSE G, RAMESHA K, et al. Reversible anionic redox chemistry in high-capacity layered-oxide electrodes[J]. Nature Materials, 2013, 12(9): 827-835. DOI:10.1038/nmat3699 |
| [21] |
HONG J, LIM H D, LEE M, et al. Critical role of oxygen evolved from layered Li-excess metal oxides in lithium rechargeable batteries[J]. Chemistry of Materials, 2012, 24(14): 2692-2697. DOI:10.1021/cm3005634 |
| [22] |
ZHU G, WEN K, LV W, et al. Materials insights into low-temperature performances of lithium-ion batteries[J]. Journal of Power Sources, 2015, 300: 29-40. DOI:10.1016/j.jpowsour.2015.09.056 |
| [23] |
WANG M, LUO M, CHEN Y, et al. Electrochemical deintercalation kinetics of 0.5Li2MnO3·0.5LiNi1/3Mn1/3Co1/3O2 studied by EIS and PITT[J]. Journal of Alloys and Compounds, 2017, 696: 907-913. DOI:10.1016/j.jallcom.2016.12.085 |
| [24] |
WEPPNER W, HUGGINS R A. Determination of the kinetic parameters of mixed-conducting electrodes and application to the system Li3Sb[J]. Journal of the Electrochemical Society, 1977, 124(10): 1569-1578. DOI:10.1149/1.2133112 |
| [25] |
YAMADA Y, IRIYAMA Y, ABE T, et al. Kinetics of electrochemical insertion and extraction of lithium ion at SiO[J]. Journal of the Electrochemical Society, 2010, 157(1): A26-A30. DOI:10.1149/1.3247598 |
| [26] |
ZHANG J N, LI Q, WANG Y, et al. Dynamic evolution of cathode electrolyte interphase (CEI) on high voltage LiCoO2 cathode and its interaction with Li anode[J]. Energy Storage Materials, 2018, 14: 1-7. DOI:10.1016/j.ensm.2018.02.016 |
| [27] |
ZHAO E, ZHANG M, WANG X, et al. Local structure adaptability through multi cations for oxygen redox accommodation in Li-rich layered oxides[J]. Energy Storage Materials, 2020, 24: 384-393. DOI:10.1016/j.ensm.2019.07.032 |
| [28] |
LU P, LI C, SCHNEIDER E W, et al. Chemistry, impedance, and morphology evolution in solid electrolyte interphase films during formation in lithium ion batteries[J]. The Journal of Physical Chemistry C, 2014, 118(2): 896-903. DOI:10.1021/jp4111019 |
| [29] |
JOW T R, ALLEN J L, MARX M, et al. Electrolytes, SEI and charge discharge kinetics of Li-ion batteries[J]. ECS Transactions, 2010, 25(36): 3-12. |
| [30] |
ASSAT G, FOIX D, DELACOURT C, et al. Fundamental interplay between anionic/cationic redox governing the kinetics and thermodynamics of lithium-rich cathodes[J]. Nature Communications, 2017, 8(1): 1-12. DOI:10.1038/s41467-016-0009-6 |
| [31] |
Van der Ven A, CEDER G, ASTA M, et al. First-principles theory of ionic diffusion with nondilute carriers[J]. Physical Review B, 2001, 64(18): 184307-184324. DOI:10.1103/PhysRevB.64.184307 |
| [32] |
MOHANTY D, KALNAUS S, MEISNER R A, et al. Structural transformation of a lithium-rich Li1.2Co0.1Mn0.55Ni0.15O2 cathode during high voltage cycling resolved by in situ X-ray diffraction[J]. Journal of Power Sources, 2013, 229: 239-248. DOI:10.1016/j.jpowsour.2012.11.144 |
| [33] |
HUA W, CHEN M, SCHWARZ B, et al. Lithium/oxygen incorporation and microstructural evolution during synthesis of Li-rich layered Li[Li0.2Ni0.2Mn0.6]O2 oxides[J]. Advanced Energy Materials, 2019, 9(8): 1803094. DOI:10.1002/aenm.201803094 |
| [34] |
NAYAK P K, GRINBLAT J, LEVI M, et al. Structural and electrochemical evidence of layered to spinel phase transformation of Li and Mn rich layered cathode materials of the formulae xLi[Li1/3Mn2/3]O2·(1-x) LiMn1/3Ni1/3Co1/3O2(x=0.2, 0.4, 0.6) upon cycling[J]. Journal of the Electrochemical Society, 2014, 161(10): A1534-A1547. DOI:10.1149/2.0101410jes |
| [35] |
JIANG W, YIN C, XIA Y, et al. Understanding the discrepancy of defect kinetics on anionic redox in lithium-rich cathode oxides[J]. ACS Applied Materials & Interfaces, 2019, 11(15): 14023-14034. |
| [36] |
SHI J L, XIAO D D, ZHANG X D, et al. Improving the structural stability of Li-rich cathode materials via reservation of cations in the Li-slab for Li-ion batteries[J]. Nano Research, 2017, 10(12): 4201-4209. DOI:10.1007/s12274-017-1489-3 |