 2021, Vol. 49
2021, Vol. 49文章信息
- 姜卓钰, 束小文, 吕晓旭, 高晔, 周怡然, 董禹飞, 焦健
- JIANG Zhuo-yu, SHU Xiao-wen, LYU Xiao-xu, GAO Ye, ZHOU Yi-ran, DONG Yu-fei, JIAO Jian
- SiC晶须增强SiCf/SiC复合材料的力学性能
- Mechanical properties of SiC whiskers reinforced SiCf/SiC composites
- 材料工程, 2021, 49(8): 89-96
- Journal of Materials Engineering, 2021, 49(8): 89-96.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2020.000806
-
文章历史
- 收稿日期: 2020-08-26
- 修订日期: 2021-06-08




2. 中国航发北京航空材料研究院 表面工程研究所, 北京 100095;
3. 陆军装备部航空军事 代表局驻北京地区航空军事代表室, 北京 100101
2. Surface Engineering Division, AECC Beijing Institute of Aeronautical Materials, Beijing 100095, China;
3. Army Aviation Representative Office in Beijing, Affiliated with the Equipment Department of People's Liberation Army Ground Force, Beijing 100101, China
近年来,随着发动机推重比的不断提高,燃烧室、涡轮与加力燃烧室等热端部件对材料的耐高温性提出了更高的要求。相比传统的高温合金材料,SiCf/SiC复合材料可以有效减轻部件质量,减少冷空气流量,显著提升工作温度,因而是制备高性能发动机高温构件的理想材料[1-2]。而SiCf/SiC复合材料在高温使用过程中,基体会形成孔洞和裂纹等缺陷,导致材料脆性大,损伤韧性不足,疲劳寿命短,难以满足下一代发动机材料疲劳性能的需求[3-5]。因此现阶段有必要探索微纳米增强体多级增韧等技术途径,开发更耐高温、更高损伤容限的SiCf/SiC复合材料。
纳米增强体作为第二增强体引入到复合材料基体中,可通过裂纹偏转及桥连等增韧机制,从多尺度提高增强相的增强效果,进一步提高复合材料的耐氧化性能、力学性能等。SiC晶须(SiCw)是一种性能优异的纳米增强体,将其引入到纤维布层间和纤维束间的脆性基体中,可有效增加裂纹扩展距离,改善微区基体的脆性,提高微区基体的韧性,从而进一步提高陶瓷基复合材料的力学性能[6-7]。同时由于纳米增强体可以阻碍基体脆性裂纹中的氧分子的流通,因此能够有效改善陶瓷基复合材料的高温耐氧化性能[8]。
Hui等[9]在Cf/SiC复合材料中引入了SiC晶须后,复合材料的性能明显提高。而采用SiC晶须或纳米纤维作为增强体时,易与SiC基体之间形成强结合,不利于发挥SiC纳米增强体的作用,因此,需要在纳米增强体表面沉积一层界面层[10]。研究者采用CVD法在纳米纤维表面沉积了热解碳(PyC)界面层,发现复合材料的性能得到明显改善[11-12]。然而PyC界面层的起始氧化温度为450 ℃,作为界面层时不利于复合材料的长时高温性能[13]。而六方的BN界面层晶体结构与PyC类似,由于其起始氧化温度较高,作为界面层时具有更加优异的抗氧化性能,因而经常被用作SiCf/SiC复合材料中的界面相[14-15]。本工作采用前驱体浸渍裂解工艺制备连续SiC纤维和SiC晶须多级增强的SiCf/SiC-SiCw复合材料,并在SiC晶须表面制备BN界面层,研究SiC晶须及BN界面层对复合材料力学性能的影响。
1 实验材料及方法 1.1 实验材料增强体纤维采用国产二代SiC纤维;在SiC纤维表面沉积BN界面层,作为纤维与基体之间的界面相;SiC晶须由浙江金刚云纳米纤维科技有限公司提供,具体参数见表 1。
| Density/(g·cm-3) | Diameter/nm | Purity/% | Mass fraction of carbon/% | Young’s modulus/GPa | Bending strength/GPa |
| 3.2 | 100-500 | ≥98 | ≤0.5 | 610-660 | 53.4 |
聚碳硅烷(Polycarbosilane, PCS)为橙黄色液体,由中国科学院化学研究所提供,固化温度为120~240 ℃。
1.2 样品制备SiCf/SiC复合材料的制备:①在SiC纤维表面涂刷PCS树脂制备预浸料;②将预浸料热压固化制备出平板实验件;③高温烧结得到多孔平板;④采用前驱体浸渍多孔平板;⑤循环步骤③和④进行致密化。循环9次制备出复合材料平板件(下同),将本试样标记为S1。
SiCf/SiC-SiCw复合材料的制备:①分别将体积分数为10%和20%SiC晶须添加到PCS中;②将混合物装入尼龙罐并放置在球磨机上球磨制备混合料浆,球磨机转速设置为160 r/min,球磨5 h得到混合料浆;③将混合料浆涂刷至SiC纤维上得到预浸料。随后的热压、高温烧结、浸渍及循环过程同S1制备步骤中②~⑤。将SiC晶须体积分数为10%和20%的试样分别标记为S2和S3。
SiCf/SiC-SiCw(BN)复合材料的制备:本样品中晶须含量、料浆制备、预浸料制备、热压固化及高温烧结方法同S2。不同的是本样品初次烧结后将样品放入化学气相沉积炉进行BN沉积,结束后再采用前驱体浸渍。随后循环高温烧结-浸渍过程进行致密化,将本试样标记为S4。
1.3 测试表征试样的密度和显气孔率采用排水法进行测试;弯曲强度采用MTS810材料实验系统进行测试,参照标准为GB/T 6569-2006;断裂韧度采用微机控制电子万能试验机C45.105进行测试,参照标准为GB/T23806-2009;拉伸强度采用Instron8801型万能材料试验机进行测试,参照标准为GJB6475-2008;压缩强度采用微机控制电子万能试验机C45.105进行测试,参照标准为GJB 6476-2008。采用Nova Nano SEM450扫描电子显微镜(SEM)对样品的微观形貌进行观察。
2 结果与讨论图 1为SiC纤维表面制备了界面层后的微观形貌和EDS图谱。可见沉积了界面层后,SiC纤维单丝之间并未粘连,SiC纤维保留了较为光滑致密的表面。由图 1插图可见,SiC纤维的直径约13 μm,其外层包裹着一层均匀的界面层,界面层与SiC纤维结合紧密,可避免界面层从纤维表面脱落。由EDS结果可以看出,该界面层为BN。

|
图 1 沉积界面层后SiC纤维的微观形貌(a)和EDS图谱(b) Fig. 1 SEM images(a) and EDS spectrum(b) of silicon carbide fibers with interphase |
将SiC晶须混入PCS前驱体制备出混合料浆,其微观形貌如图 2(a)所示。由图 2(a)可见混合料浆中SiC晶须亮度较高。由高倍放大形貌可见,高亮度SiC晶须具有较好的线性均匀性,其直径约为100 nm。图 2(b)为混合料浆的面扫描和EDS能谱分析结果,可见混合料浆中C元素与Si元素均匀分布,表明SiC晶须在混合料浆中分散得较为均匀。

|
图 2 混合料浆的SEM照片(a)和面扫描及EDS分析结果(b) Fig. 2 SEM images (a), mapping and EDS spectrum (b) of slurry with SiC whiskers |
以图 1所示沉积界面层后的SiC纤维为连续增强体,采用PCS前驱体和图 2所示的混合料浆制备复合材料S1和S2。对复合材料进行物理性能测试,结果如表 2所示。由表可见,S1和S2样品的密度和孔隙率较为接近。但在S2样品中引入了SiC晶须后,复合材料中基体的体积有所增加,导致S2样品的纤维体积分数相比于S1有所降低。
| Sample | Volume fraction of SiC whisker/% | Apparent density/ (g·cm-1) | Volume density/ (g·cm-1) | Apparent porosity/% | Volume fraction of fiber/% |
| S1 | 2.52 | 2.42 | 4.01 | 24.34 | |
| S2 | 10 | 2.47 | 2.39 | 3.52 | 21.48 |
| S3 | 20 | 2.50 | 2.40 | 3.86 | 21.56 |
| S4 | 10 | 2.38 | 2.26 | 5.13 | 21.98 |
表 3为S1和S2样品的力学性能结果。可见相比于S1样品,S2样品的压缩强度提高了14.6%,但其拉伸强度、弯曲强度及断裂韧度均有所降低。为了验证引入SiC晶须对SiCf/SiC复合材料的压缩强度的影响规律,将混合料浆中SiC晶须的含量提高至20%,制备出S3复合材料,其物理性能如表 2所示。对S3样品的压缩强度进行测试,结果如图 3所示。可见当SiC晶须含量提高至20%时,复合材料压缩强度进一步提高至673.9 MPa,比S1样品高22.6%。
| Sample | Compressive strength/ MPa | Tensile strength/MPa | Flexural strength/MPa | Fracture toughness/(MPa·m1/2) |
| S1 | 549.70 | 400.15 | 767.86 | 20.85 |
| S2 | 629.82 | 363.50 | 735.58 | 18.66 |

|
图 3 S1~S3样品的压缩强度 Fig. 3 Compressive strengths of S1-S3 |
图 4为S1~S3样品压缩断口的微观形貌。由图 4(a)可见,S1样品受压缩载荷产生裂纹后,横向纤维束层间的基体层发生断裂,层内纤维延续性较好,基体断口较为平整。由图 4(b)可见S2样品压缩断口中出现多层横向纤维断口,基体断口高低起伏较为明显。同样,在S3样品断口中,不同层断裂的横向纤维和断口处基体的起伏更明显(图 4(c))。

|
图 4 S1(a), S2(b)和S3(c)样品的压缩断口 Fig. 4 Compressive fracture morphologies of S1(a), S2(b) and S3(c) |
当复合材料基体中受载荷作用形成微裂纹时,分布在基体中的SiC晶须与基体发生脱粘拔出(图 5(a))、桥连(图 5(b))及裂纹偏转(图 5(c))等作用,使得裂纹尖端应力松弛,减缓了裂纹的扩展速度,延长扩展路径,消耗更多的能量,从而使复合材料具有更高的强度[16-18]。结合图 4可见,S1样品基体中未添加SiC晶须,因此复合材料基体产生裂纹后,在层内的基体中迅速扩展,导致复合材料失效。而在S2和S3样品中,裂纹在复合材料基体中扩展时一般较难穿过晶须,更容易绕过晶须并沿晶须表面扩展,即发生了裂纹偏转作用。这种偏转作用使基体中的裂纹发生层间扩展,避免了层内迅速扩展,延长扩展路径,降低了裂纹扩展时的拉应力,增加了裂纹扩展过程中的能量消耗[19];当复合材料出现较大裂纹时,SiC晶须的拔出和桥连作用也会增加裂纹扩展时的能量消耗,从而使得复合材料的压缩强度明显提高。

|
图 5 SiC晶须增强复合材料中的晶须拔出(a)、桥连(b)和裂纹偏转(c) Fig. 5 SEM images for whisker pullout (a), bridging (b) and crack deflection (c) of SiC whisker reinforced composites |
同时,在S1样品压缩断口的微观形貌(图 4(a))中,明显可见与基体完全脱粘的SiC纤维单丝。S2样品中SiC纤维被基体包裹,但SiC基体与纤维之间存在明显间隙(图 4(b)中箭头所示)。由图 4(c)可见S3样品中纤维被基体完全包裹,且基体与纤维的结合较为紧密,因此在S1~S3样品中基体与纤维的结合强度依次增加。
图 6为S1和S2样品在拉伸过程中典型的应力-应变曲线。由图可见S2样品在弹性变形阶段的曲线斜率明显大于S1样品,表明晶须的引入使得S2样品的模量有所提高[20-21]。在复合材料中引入SiC晶须后,其模量(E)可通过式(1)计算得出[12]。
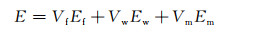
|
(1) |

|
图 6 S1和S2样品的应力-应变曲线 Fig. 6 Stress-strain curves of S1 and S2 |
式中:Vf,Vw和Vm分别代表纤维、晶须和基体的体积分数;Ef,Ew和Em分别代表纤维、晶须和基体的模量。
结合表 2可知S2样品的纤维体积分数明显小于S1样品,因此S2复合材料模量的提高是由于复合材料基体中引入了高模量SiC晶须。
图 7为S1和S2样品拉伸断口的微观形貌。可见两个样品的断口处有不同程度的纤维拔出。S1样品拉伸断口的纤维拔出较长,而S2复合材料的拉伸断口较为平整且纤维拔出较短,这与图 6中S2的应变较小相符。由此可见相比于S1样品,S2样品倾向于脆性断裂模式;同时,由表 2可见S2样品中连续增强纤维的体积分数有所降低,因此其拉伸强度、断裂韧度及弯曲强度有所降低。这种断裂模式的变化可能是由于在复合材料基体中直接引入SiC晶须后,晶须与SiC基体形成强结合,进而使SiC基体与连续增强纤维的结合强度较高,增加了载荷由基体向增强体的传递效率,导致连续增强纤维拔出困难[12, 20]。因此可在SiC晶须表面制备BN界面层,弱化基体与纤维的界面作用[22-23]。

|
图 7 S1(a)和S2(b)复合材料的拉伸断口形貌 Fig. 7 Tensile fracture morphologies of S1(a) and S2(b) |
沉积BN后SiC晶须的直径约为222.5 nm(图 8(a))。可见沉积BN界面层后晶须表面光滑且无明显凸起,SiC晶须直径有明显增加。由图 8(b)能谱分析结果可见,除Si和C元素外,SiC晶须表面明显含有B和N元素,表明SiC晶须表面成功制备了BN界面层。

|
图 8 沉积BN后SiC晶须的SEM(a)和EDS图谱(b) Fig. 8 SEM image (a) and EDS spectrum (b) of SiC whisker with BN interphase |
S4样品的基本物理性能如表 2所示。相比于S2样品,S4样品的纤维体积分数与其接近,但S4样品的密度有所降低,孔隙率略有增加,这是由于在BN沉积过程中,在S4样品内部局部区域形成了体积较小的封闭孔。
图 9为S4和S2样品的力学性能对比,可见S4样品的拉伸强度、弯曲强度及断裂韧度分别为414.0, 800.3 MPa和22.2 MPa·m1/2,比S2样品分别提高了13.9%, 8.8%和19.0%。由S4样品微观形貌(图 10)可见S4样品拉伸断口起伏明显,纤维拔出较长。表明BN界面层有效地缓解了增强体与基体间较强的结合作用,更有利于纤维拔出,因此在典型的应力-应变曲线中(图 11),S4样品的最大载荷和断裂应变相比于S2样品均有明显提高。

|
图 9 S2和S4样品的力学性能对比 Fig. 9 Comparison of mechanical properties of S2 and S4 |

|
图 10 S4样品的拉伸断口形貌 Fig. 10 Tensile fracture morphologies of S4 |

|
图 11 S2和S4样品的应力-应变曲线 Fig. 11 Stress-strain curves of S2 and S4 |
(1) SiC晶须增强复合材料受外界载荷产生裂纹时,晶须的拔出、桥连及裂纹偏转等作用增加了裂纹在基体中传递时的能量消耗,使SiCf/SiC复合材料的压缩强度有明显提高,当引入体积分数为20%的SiC晶须时,复合材料压缩强度提高了22.6%,可达673.9 MPa。
(2) SiC晶须表面制备BN界面层后,可以降低增强体与基体的结合强度,有利于SiC晶须和纤维的拔出。因此S4样品的拉伸强度、弯曲强度及断裂韧度分别为414.0,800.3 MPa和22.2 MPa·m1/2,相比于S2样品分别提高了13.9%,8.8%和19.0%。
| [1] |
焦健, 陈明伟. 新一代发动机高温材料-陶瓷基复合材料的制备、性能及应用[J]. 航空制造技术, 2014(7): 62-69. JIAO J, CHEN M W. New generation of high temperature material for engine preparation, property and application of ceramic matrix composites[J]. Aeronautical Manufacturing Technology, 2014(7): 62-69. DOI:10.3969/j.issn.1671-833X.2014.07.007 |
| [2] |
刘彦杰, 马武军, 王松. 陶瓷基复合材料火箭发动机推力室研究进展[J]. 宇航材料工艺, 2007(4): 1-4. LIU Y J, MA W J, WANG S. Research progress in ceramic matrix composites rocket thrusters[J]. Aerospace Materials & Technology, 2007(4): 1-4. DOI:10.3969/j.issn.1007-2330.2007.04.001 |
| [3] |
RUGGLES W M, BOUCHER N, PRZYBYLA C. Fatigue of three advanced SiC/SiC ceramic matrix composites at 1200 ℃ in air and in steam[J]. International Journal of Applied Ceramic Technology, 2018, 15(1): 3-15. DOI:10.1111/ijac.12773 |
| [4] |
马晓康, 殷小玮, 范晓孟, 等. 碳化硅陶瓷基复合材料的自愈合及结构吸波一体化研究进展[J]. 航空材料学报, 2018, 38(5): 1-9. MA X K, YIN X W, FAN X M, et al. Progress on self-healing and structure-wave absorbing integration of silicon carbide ceramic matrix composites[J]. Journal of Aeronautical Materials, 2018, 38(5): 1-9. |
| [5] |
WEI F, ZHANG L, LIU Y, et al. Fabrication of(SiCf+SiCw)/SiC composites by CVI combined with tape casting[J]. Ceramics International, 2015, 41(8): 9995-9999. DOI:10.1016/j.ceramint.2015.04.081 |
| [6] |
CHU Y H, FU Q G, LI H J, et al. Effect of SiC nano-wires on the mechanical and oxidation protective ability of SiC coating for C/C composites[J]. Journal of the American Ceramic Society, 2012, 95: 739-745. DOI:10.1111/j.1551-2916.2011.04979.x |
| [7] |
HUA Y, ZHANG L, CHENG L, et al. Microstructure and high temperature strength of SiCw/SiC composites by chemical vapor infiltration[J]. Materials Science and Engineering: A, 2010, 527(21/22): 5592-5595. |
| [8] |
DAI J, SHA J, SHAO J, et al. In-situ growth of SiC nanostructures and their influence on anti-oxidation capability of C/SiC composites[J]. Corrosion Science, 2017, 124: 71-79. DOI:10.1016/j.corsci.2017.05.008 |
| [9] |
HUI M, WANG H, HUI D, et al. Strength and toughness improvement in a C/SiC composite reinforced with slurry-prone SiC whiskers[J]. Ceramics International, 2014, 40(9): 14099-14104. DOI:10.1016/j.ceramint.2014.05.141 |
| [10] |
BESMANNA D P S, KUPPA E R, SHANMUGHAMA S, et al. Fiber-matrix interfaces in ceramic composites[R]. MRS Proc, 1995, 458: 147.
|
| [11] |
秦浩, 董绍明, 胡建宝, 等. PIP法制备SiC纳米线增强SiCf/SiC复合材料及其力学性能[J]. 硅酸盐学报, 2016, 44(10): 1532-1537. QIN H, DONG S M, HU J B, et al. Effect of SiC nanowires on mechanical properties of PIP-SiCf/SiC composites[J]. Journal of the Chinese Ceramic Society, 2016, 44(10): 1532-1537. |
| [12] |
HE F, LIU Y S, TIAN Z, et al. Improvement of the strength and toughness of Cf/SiC composites via chemical vapor infiltration-grown SiC nanowire interphases[J]. Ceramics International, 2018, 44(2): 2311-2319. DOI:10.1016/j.ceramint.2017.10.197 |
| [13] |
吕晓旭, 姜卓钰, 周怡然, 等. BN/SiC复合界面层对SiC纤维和PIP-Mini复合材料力学性能的影响[J]. 无机材料学报, 2020, 35(10): 1009-1104. LV X X, JIANG Z Y, ZHOU Y R, et al. Effect of BN/SiC multilayered interphases on mechanical properties of SiC fibers and minicomposites by PIP[J]. Journal of Inorganic Materials, 2020, 35(10): 1009-1104. |
| [14] |
WING B L, HALLORAN J W. Subsurface oxidation of boron nitride coatings on silicon carbide fibers in SiC/SiC ceramic matrix composites[J]. Ceramics International, 2018, 44(14): 17499-17505. DOI:10.1016/j.ceramint.2018.06.221 |
| [15] |
LV X, ZHE Q, YANG J H, et al. The effect of boron nitride interphase on the thermal stability of SiC fibers[J]. Journal of Alloys and Compounds, 2020, 844: 156193. DOI:10.1016/j.jallcom.2020.156193 |
| [16] |
梁飞, 赵婧. 晶须增韧陶瓷基复合材料匹配原则及影响因素[J]. 陶瓷, 2018(9): 59-64. LIANG F, ZHAO J. Matching principle and influencing factors of whisker toughened ceramic matrix composites[J]. Ceramics, 2018(9): 59-64. DOI:10.3969/j.issn.1002-2872.2018.09.011 |
| [17] |
CHEN N Q, CHENG L F, LIU Y S, et al. Microstructure and properties of SiCw/SiC composites prepared by gel-casting combined with precursor infiltration and pyrolysis[J]. Ceramics International, 2018, 44: 969-979. DOI:10.1016/j.ceramint.2017.10.031 |
| [18] |
PU D M, CHEN P J, XIAO P, et al. Oxidation and thermal cycling behavior of C-AlPO4 and SiC whisker comodified mullite deposited on SiC-C/SiC composites[J]. Surface & Coatings Technology, 2020, 400: 126201. |
| [19] |
LI L, ZHANG C, ZHANG C, et al. Compressive strength and damage mechanisms of 2D-C/SiC composites at high temperatures[J]. Ceramics International, 2018, 44: 14026-14031. DOI:10.1016/j.ceramint.2018.04.255 |
| [20] |
HU J B, DONG S M, FENG Q, et al. Tailoring carbon nanotube/matrix interface to optimize mechanical properties of multiscale composites[J]. Carbon, 2014, 69(4): 621-625. |
| [21] |
WANG S, ZHANG C, LIANG R, et al. Load-transfer in functionalized carbon nanotubes/polymer composites[J]. Chemical Physics Letters, 2008, 457(4): 371-375. |
| [22] |
SREEJITH K, VIPIN V, SUBRAMANIA S, et al. A comparative study on Cf/PyC/SiC minicomposites prepared via CVI process for hypersonic engine application[J]. International Journal of Applied Ceramic Technology, 2018, 15(5): 1110-1123. DOI:10.1111/ijac.12898 |
| [23] |
NASLAIN R R, PAILLER R J F, LAMON J L. Single and multilayered interphases in SiC/SiC composites exposed to severe environmental conditions[J]. International Journal of Applied Ceramic Technology, 2010, 7(3): 263-275. |